J.ophthalmol.(Ukraine).2022;5:54-64.
|
http://doi.org/10.31288/oftalmolzh202255464 Received: 03.08.2022; Accepted: 22.08.2022; Published on-line: 27.10.2022 A case of acute posterior multifocal placoid pigment epitheliopathy: clinical analysis and differential diagnosis N. V. Konovalova 1, N. I. Khramenko 1, O. V. Guzun 1, S. B. Slobodianyk 1, O. V. Kovtun 2 1 SI "The Filatov Institute of Eye Diseases and Tissue Therapy of the NAMS of Ukraine) 2 Odesa National Medical University Odesa (Ukraine) TO CITE THIS ARTICLE: Konovalova NV, Khramenko NI, Guzun OV, Slobodianyk SB, Kovtun OV. A case of acute posterior multifocal placoid pigment epitheliopathy: clinical analysis and differential diagnosis. J.ophthalmol.(Ukraine).2022;5:54-64. http://doi.org/10.31288/oftalmolzh202255464
Background: Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is a rare inflammatory disease, and the differential diagnostic assessment is essential for its adequate treatment. Purpose: To perform clinical analysis and differential diagnosis for a case of APMPPE. Material and Methods: We present a case of a 26-year-old woman with APMPPE. The patient underwent an eye examination which included visual acuity assessment, refractometry, tonometry, perimetry, biomicroscopy, direct and indirect ophthalmoscopy, optical coherence tomography (OCT), fundus photography, and fluorescein angiography (FA). Endonasal electrophoresis was conducted in a routine manner using azithromycin, a macrolide antibiotic. Results: OCT found numerous flat yellowish-white and grey-pink lesions in the posterior pole of the fundus at the level of the retinal pigment epithelium and choriocapillary layers. FA showed irregularly shaped non-fluorescent lesions, which began to exhibit faint fluorescence a few minutes after the dye injection. The differential diagnosis should include serpiginous choroiditis, Vogt-Koyanagi-Harada syndrome, geographical choroiditis, acute retinal pigment epitheliitis (Krill disease), and choroidal neoplasm. Conclusion: Acute posterior multifocal placoid pigment epitheliopathy is an acute-onset disease. If the disease is suspected, a thorough history is essential, and the differential diagnosis should be performed for early treatment and referral to an appropriate specialist. Patients with this disease should be followed since recurrences can occur. Keywords: acuteposterior multifocal placoid pigment epitheliopathy, differential diagnosis, treatment
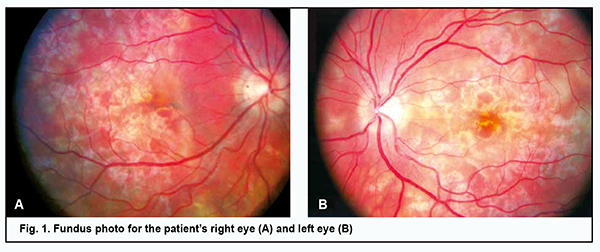
Introduction Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is a rare inflammatory disease, classified as a part of the spectrum of the white dot syndromes, a terminology presently revised. Today it is classified within the group of choriocapillaritis diseases [1]. The disorder was first described by Gass in 1968 as multiple, discrete, cream-coloured lesions located in the posterior pole of three young women complaining of painless visual reduction [2]. The disease is found equally between men and women, with a male to female ratio of 1.2:1, and is more common in Caucasian people [3]. Data combined from published case reports identified 27 years as the mean age of onset of the disorder, with a range between the first and seventh decade, and over three quarters of cases occurring between 16 and 40 years of age [1, 3, 4]. Gass considered the retinal pigment epithelium (RPE) to be the primary site of inflammation, with RPE edema leading to an impaired cell supply and inactivation of metabolism in the retina. Others, however, believe that APMPPE primarily affects the choriocapillaris and inner choroid [5]. Multimodal imaging features in APMPPE support the most likely primary inflammatory involvement of the choriocapillaris with secondary photoreceptor disruption, resulting in the characteristic distinctive clinical phenotype of the disease [1]. However, the exact underlying etiologic mechanism is unknown, although an immune driven nature has been hypothesized based on the following associations. The disease is often preceded by a viral prodrome. APMPPE develops after a flu-like condition in 30% of patients. In addition, cases of APMPPE developing after manifestation of adenovirus infection have been reported [3, 6]. Numerous cases of APMPPE have been reported in patients with a positive tuberculosis immunological test. Cases of APMPPE associated with epidemic parotitis and Lyme disease have been cited [1, 7, 8]. The disease has also been linked with pre-existing autoimmune and autoinflammatory diseases, including psoriasis, sarcoidosis, erythema nodosum, polyarthritis nodosum, ulcerative colitis, eczema, diabetes mellitus, and Wegener’s granulomatosis, and cases of patients developing such conditions after being diagnosed with APMPPE have also been described [1, 9-11]. An increased incidence of both class I HLA antigen (HLA-B7) and class II HLA antigen (HLA-DR2) has been reported in APMPPE patients [12]. There are reports of APMPPE occurring in healthy individuals after immunization with vaccines [13]. The immune driven nature is supported by the development of the disease after immunization with vaccines, including hepatitis B, varicella, polio, and tetanus, suggesting a possible interplay between vaccines and specific T-cell receptors of the host immune system [1, 14, 15]. Rarely, APMPPE may be the first manifestation of vasculitis of the central nervous system [16]. Today there is an agreement among researchers that choroidal vasculitis with occlusion of precapillary arterioles and ischemia in individual choroidal lobes is an initial event in the pathogenesis of APMPPE, while a secondary event is the development of subsequent ischemic RPE lesions [17]. Clinical manifestations Patients complain of bilateral sudden and painless visual loss, photopsias and paracentral scotomata. In most cases patients have bilateral disease with the second eye affected within a few days or weeks following the fellow eye. The lesions tend to gradually fade over few weeks, but new lesions may appear up to three weeks following the onset, coexisting with older lesions. Ophthalmoscopic manifestations include multifocal, yellowish creamy, subretinal, placoid lesions, located posteriorly to the equator, generally within the posterior pole. The disease may be associated with minimal inflammatory reaction in the vitreous cavity, usually in the absence of anterior uveitis. Papillitis and retinal vasculitis may be present [1]. Algahtani and colleagues [18] reported cerebral vasculitis as the most common neurological complication, affecting 50% of the patients, following by isolate headaches in 26.8%. Other neurological complications include cavernous sinus thrombosis, meningoencephalitis, aseptic meningitis, stroke or transient ischemic attack, seizures, sixth-cranial-nerve palsy, and transient hearing loss. Instrumental diagnostic techniques include imaging procedures. Serial fundus photography from acute onset to the stage of healing is a useful tool that allows documenting the evolution of polymorphic lesions and can also help in detecting the development of potential complications including development of choroidal neovascularization manifesting as hemorrhage contiguous to the scar. In the early phases of fundus fluorescein angiography (FA), active APMPPE lesions show hypofluorescence which is likely to represent poor perfusion or delayed filling of the choriocapillaris. The late hyperfluorescence is explained by the profound ischaemia of the outer retina which could produce reactionary hyperpermeability and exudation from retinal vessels. In the late phases of the disease, there are signs of RPE atrophy. On indocyanine green angiography (ICGA) active placoid lesions appear as hypofluorescent spots across all phases of the exam, likely due to poor perfusion of the choriocapillaris. On spectral-domain optical coherence tomography (SDOCT), the placoid lesions in the acute stage appear as disruption of the outer retinal and ellipsoid zone, with the presence of hyperreflective material at the level of the outer retinal layers and RPE. Over the course of resolution of the placoid lesions, the hyperreflectivity of the outer retina disappears, resulting in either partial restoration of its appearance or focal areas of photoreceptors/RPE atrophy [19, 20]. Four distinct OCT phases of APMPPE have been described [21]. In the first phase, there is a dome-shaped elevation with disruption of the photoreceptor junction that flattens soon after. Two weeks later, the second phase occurs and is characterized by the detection of a distinct separation between the photoreceptor junction and the RPE. The third stage, occurring six weeks after disease onset, is characterized by increased RPE hyperreflectivity and union of the RPE and photoreceptor junction. However, focal areas of photoreceptors/RPE atrophy can develop. The resolution phase starts at three months post disease onset, with the reformation of two distinct visible layers of photoreceptors and RPE. Focal areas of photoreceptors/RPE atrophy can, however, persist. A case of APMPPE with exudative retinal detachment, simulating acute Vogt-Koyanagi-Harada (VKH) disease has been reported [22]. In patients with APMPPE, optical coherence tomography angiography (OCT-A) demonstrates areas of decreased flow/flow void at the level of the choriocapillaris [23]. These areas of flow deficit correlate closely with the ischemic lesions manifesting as early hypofluorescent on FA and hypofluorescent throughout the exam on ICGA. Although the diagnosis of APMPPE is based on clinical features and multimodal imaging findings, an infectious etiology and associated systemic causes need to be ruled out. Laboratory investigations for tuberculosis, syphilis and autoimmune disorders should be carried out. In case of significant headaches and on suspicion of neurologic involvement, patients must be investigated with brain magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) examination. Although the original description categorized APMPPE as a self-limited condition with a good prognosis, the reality is that the disease can be recurrent and result in significant visual loss and CNS lesions. The purpose of the study was to perform clinical analysis and differential diagnosis for a case of acute posterior multifocal placoid pigment epitheliopathy. Material and Methods We present a case of a 26-year-old woman with APMPPE who was examined, treated and followed up at the Department for General Ocular Pathology, the Filatov Institute of Eye Diseases and Tissue Therapy. Functional diagnostic assessment and electrophysiological examination were carried out at Ocular Function Assessment Laboratory, and imaging studies were carried out at Laboratory for Studies on the Biological Effect and Use of Lasers. The patient underwent an eye examination which included visual acuity assessment, refractometry, tonometry, perimetry, biomicroscopy, direct and indirect ophthalmoscopy, fundus photography, and FA. In addition, she underwent ophthalmic rheography (ORG) with Reocom (KHAI-Medika, Kharkiv, Ukraine), the computerized rheography apparatus. ORG included measurements of ocular pulse blood filling (OPBF, expressed as RQ, ‰ rheographic coefficient) and vascular tone (expressed as alpha/T percentage index). Vascular tones in large-caliber vessels and small-caliber vessels were determined using high-frequency and low-frequency characteristics of the differential ROG curve. Volumetric ocular blood flow velocity was determined for the rising portion of ROG curve and assessed in Ohm per second. Visual evoked potentials (VEP) were recorded using a RetiScan unit (Roland Сonsult, Germany). Central visual fields were investigated by automated static perimetry using program 30-1 of the Humphrey Field Analyzer (HFA). The patient was examined during the acute phase of APMPPE (at presentation) and during the remission of the disease. Endonasal electrophoresis was performed using the following procedure. Nasal gauze packing soaked with 2 ml of 2% calcium chloride and 1 ml of an antibacterial drug (Azyter® 250 mg) is used as the active electrode. The anode is positive. The current increases in a stepwise manner from 0.3 mA to 0.5 mA, 0.8 mА and 1 mА at 3, 5, 8 and 10 minutes, respectively. The dispersing electrode fitted with a hydrophilic gasket is placed in the shoulder-and-neck area. Azyter® (azithromycin) is a broad-spectrum macrolide antibiotic of the azalide subclass, which has been shown to display high bactericidal activity. It exerts bacteriostatic and bactericidal effects by binding to the 50S ribosomal subunit of bacteria, thereby preventing peptide translocation, and subsequent protein synthesis. The study was conducted in accordance with applicable local laws and the principles stated in the Declaration of Helsinki and the European Convention on Human Rights and Biomedicine. Informed consent was obtained for the use of personal data from medical records. Results A 26-year-old woman presented to the clinic complaining of reduced vision, central and paracentral scotoma, metamorphopsia and photopsia in the left eye. She had bilateral disease with the second eye affected within a few days following the fellow eye. Two weeks before presentation, she had had an acute respiratory viral infection without raised temperature. She reported being myopic since childhood. On examination, her uncorrected visual acuity OD was 0.1 but corrected to 0.4 with -3.0 D of sphere. The anterior segment OD was unremarkable. The optic disk was pale pink with sharp margins. Yellow-greyish and cream-colored, confluent lesions with poorly defined borders were seen in the posterior segment. High retinal pigment epithelial and neuroepithelial detachments were present in the macula and along the superior temporal arcade (Fig. 1A). The peripheral fundus was unremarkable. The retinal arterioles were tortuous, but the arteriolar caliber was normal.
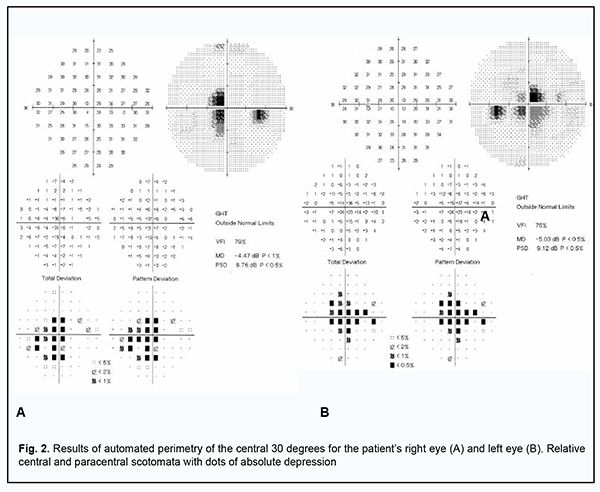
The uncorrected visual acuity OS was 0.1 but corrected to 0.35 with 3.0 D of sphere. The anterior segment OS was unremarkable. Dispersed suspended solid particles were seen in the preretinal vitreous space. The optic disk OS was pale pink with sharp margins. The retinal arterioles were tortuous, but the arteriolar caliber was normal. In the posterior segment, there were yellow-white, cream-colored or pink lesions with poorly defined borders and small edema close to the lesions (Fig. 1B). Retinal pigment epithelial and neuroepithelial detachments were present in the macula and along the superior temporal arcade. The peripheral fundus was unremarkable. The visual fields showed enlargement of the blind spots and isolated relative central and paracentral scotomata (Fig. 2).
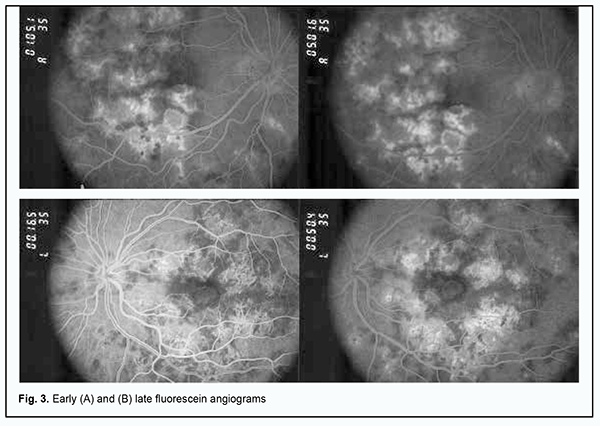
Lesions in both eyes showed hypofluorescence in the early phases of FA. The degree of hypofluorescence gradually increased with time, with the formation of areas of RPE atrophy with mildly redistributed pigment, and lesions of various severities observed in the fundus (Fig. 3A).
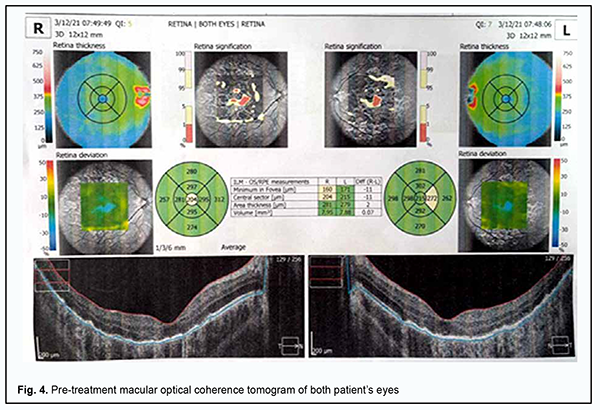
In the late phase of FA, hyperfluorescent regions could be seen throughout the fundus, and hyperfluorescent pinpoint changes could be observed in areas of serous retinal detachment. Hyperfluorescent optic disk was noted in the late phase of FA (Fig. 3B). In both eyes, OCT showed hyperreflective lesions at the level of photoreceptors, destruction of the outer photoreceptor and RPE layers, foci of proliferation and RPE detachment (Fig. 4).
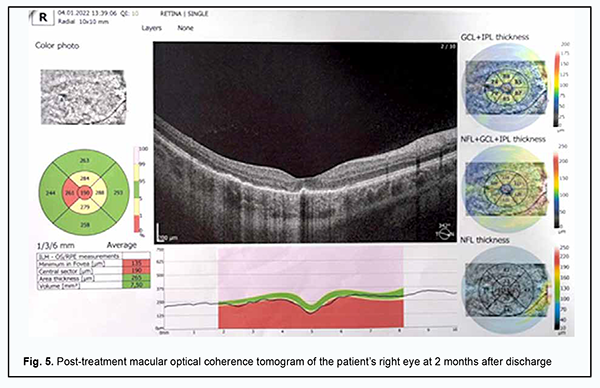
Mean pattern visual evoked potential (PVEP) P100 amplitude for a 1-degree check size was 4.8 μV in the right eye and 5.6 μV in the left eye, which was about half of normal amplitude, and PVEP P100 latency ranged from 128 ms to 132 ms, which was 30% longer than normal latency. Mean pattern visual evoked potential (PVEP) P100 amplitude for a 15-minute check size was 5.0 μV in the right eye and left eye, which was about half of normal amplitude, with PVEP P100 latency of 106 ms and 120 ms for the left and right eyes, respectively. These data demonstrate abnormalities in the right and left visual pathways, which was confirmed by 60% and 20% reductions in flash VEP amplitudes and 15% and 28% prolongations in flash VEP latencies in the right eye and left eye, respectively. Scotopic electroretinograms (ERG’s) were normal. In the maximal ERG, the a-wave represents the function of the peripheral photoreceptors and was normal. In addition, the b-wave is the measure of the activity of peripheral bipolar cells and Müller cells, and showed a 20% reduction in amplitude from normal and normal latency. The photopic ERG reflects mostly central retinal activity. The photopic ERG b-wave showed 15-20% latency prolongations and 20% amplitude reductions in both eyes, reflecting a decreased functional activity of the bipolar cells and Müller cells of the central retina. The ophthalmic rheography study showed 30% and 18% reductions in RQ (‰) in the right eye and left eye, respectively, and moderately increased vascular tones in large-caliber vessels. The patient was diagnosed with a bilateral acute posterior multifocal placoid pigment epitheliopathy and moderate myopia based on the clinical symptoms and imaging studies. She was administered 10 sessions of transorbital electrophoresis with Azyter®, non-steroidal anti-inflammatory and antiviral drugs, and vascular and metabolic therapy. Associations of APMPPE with autoimmune and autoinflammatory disorders have been reported [7, 10, 17, 18]. Therefore, antibacterial therapy was administered due to findings of bilateral retinal inflammation. At discharge (treatment day 10), the patient noted an improvement in vision, and her visual acuity with correction was 0.5 OD and 0.4 OS. One month later, she had no complaints of vision loss, and her visual acuity with correction was 0.8 OD and 0.85 OS. On eye examination in both eyes, the anterior segment was unremarkable and the media were transparent. The optic disk was pale pink with sharp margins. Ophthalmoscopy showed polymorphic lesions with significantly reduced edema adjacent to them in the posterior segment. The peripheral fundus was unremarkable. The patient was taking Nutrof® Forte (a dietary supplement containing vitamins, antioxidants, lutein, zeaxanthin, omega-3 fatty acids, and Vitis Vinifera extract (Resveratrol)) for a month. She missed scheduled follow-up visits to the institute, but was examined by a local ophthalmologist two months after discharge. Her visual acuity when corrected with -3.0 D of sphere improved to 0.9 in the right eye and 1.0 in the left eye. The visual fields showed no relative scotoma. The patient showed OCT evidence of improved APMPPE (Fig. 5) with treatment with anti-inflammatory, vascular and metabolic drugs.
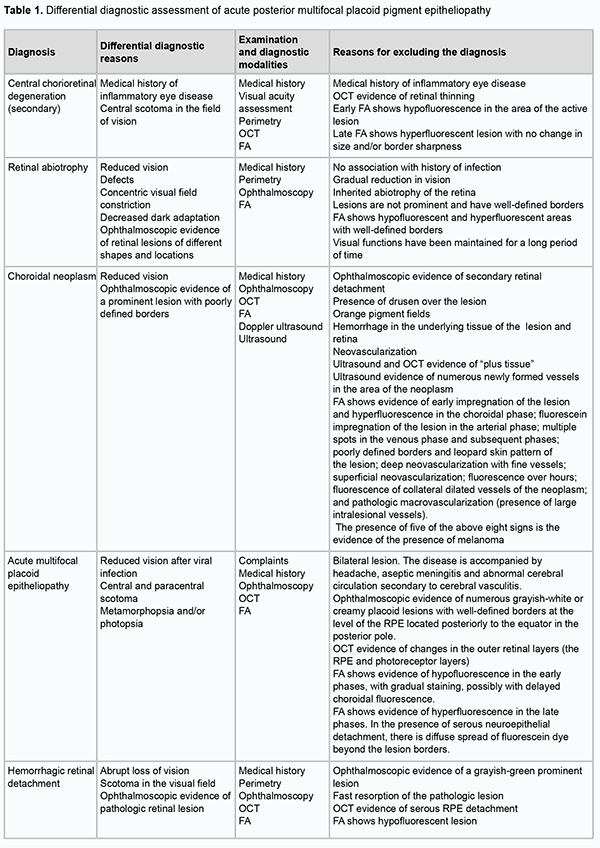
Discussion In patients with APMPPE, typical fundus changes may be accompanied by anterior uveitis, optic edema, periphlebitis [24], serous retinal detachment, and vascular occlusion. The disease may be accompanied by neurological symptoms (headache), aseptic meningitis [25], and abnormal cerebral circulation due to cerebral vasculitis [17]. Large ischemic strokes can occur and have been reported to result in patient death [26]. In cases with ocular lesions only, visual acuity typically restores back to normal within weeks to months after disease onset, although permanent paracentral scotomata may persist [27]. Choroidal neovasculariation is a rare potential late complication of APMPPE. The differentiatial diagnosis of APMPPE should include primarily serpiginous choroiditis, because ophthalmoscopic and angiographic findings in eyes with APMPPE may be similar to those in eyes with acute serpiginous choroiditis. Serpiginous choroiditis may be classified as a placoid epitheliopathy. The disease commonly affects young and middle-age individuals, displays an acute or chronic inflammatory course with recurrences, the primary lesions being in the choriocapillaris and RPE, and the secondary lesions, in the other parts of the retina. Patients with acute onset of serpiginous choroiditis complain of floaters, blurry vision and vision loss. The disease develops more slowly than APMPPE, and patients have angiographic evidence of apparent atrophic changes in choriocapillaris and choroidal vessels. The characteristic biomicroscopic findings include inflammatory cells in the posterior vitreous. Funduscopic examination demonstrates flat, greyish-white confluent lesions at the level of the RPE. A few months after disease onset, geographical patches of choroidal atrophy emerge at the sites of active lesions. In approximately one half of patients, varying amounts of grey-white tissue (fibrous metaplasia of the RPE) develop within the area of chorioretinal atrophy, resembling the picture of degeneration secondary to tuberculous choroiditis. Subretinal neovascularization and hemorrhage may result. Retinal and optic disk vessels are typically not altered. The visual field demonstrates absolute and/or relative scotomata corresponding to the geographic lesion. In chronic cases, early disease is typically not associated with vision loss, and frequently remains unidentified. In serpiginous choroiditis, lesions are located peripapillary or juxtapapillary, occasionally a serous RPE detachment may occur. At intervals varying from weeks to months, the patient is subject to recurring episodes of varying disease activity. Ophthalmoscopy finds fresh lesions involving new and contiguous areas of choroidal atrophy and macular edema. In addition, it reveals numerous old chorioretinal scars in the peripheral fundus. Inflammatory cells are seen in the posterior vitreous, evidencing the progression of the process. Inflammatory cell reaction is present in the posterior vitreous in approximately one-third of cases during the active phase of the disease. Examination of the opposite eye often reveals an area of chorioretinal scarring adjacent to the optic disc. In serpiginous choroiditis, there is angiographic evidence of atrophy of choriocapillaris and choroidal vessels. During the early phases of angiography, the acute gray-white lesions appear nonfluorescent due to destruction of the choriocapillaris, and later they show evidence of staining that usually begins at the border of the lesion and spreads centrally. As fluorescein diffuses from the neighboring choriocapillaris, the atrophic lesions show progressive staining from the margins centrally. The detection of hyperfluorescence at the borders of lesions during the late phases of angiography may be indicative of the activity of the process. It is difficult to differentiate serpiginous choroiditis from tuberculous choroiditis solely on the basis of ophthalmoscopy. Specific tests like X-ray of the kidneys and Mantoux test will be helpful for the differentiation. While differentiating APMPPE from multifocal chorioretinitis, it is important taking into account that, in chorioretinitis, prominent inflammatory lesions are located in the choroid. In the advanced phases, multifocal pigment epitheliopathy can be differentiated from tapetoretinal abiotrophy, because there is evidence of changes in electrophysiological characteristics in the latter disease, but not in the former disease [28, 29]. In our patient with APMPPE, the ERG characteristics were slightly different from normal and yellow-greyish and cream-colored, confluent lesions with poorly defined borders were seen in the posterior segment. In serpiginous choroiditis, lesions are located peripapillary or juxtapapillary, but in our patient, lesions were located in the posterior segment. In addition, there was a significant difference in color between the brownish pink-cream-colored lesions in our patient and grey-black lesions characteristic for serpiginous choroiditis. However, our visual evoked potential records from the patient revealed abnormal function of the visual pathway, demonstrating the necessity for the follow-up examination, including an examination by the neurologist. Acute retinal pigment epitheliitis is another disease to consider in the differential diagnosis of APMPPE. The etiology of the disease is unknown. It has been hypothesized that the development of the disease is linked to viral replication in the RPE. Acute RPE inflammation is an initial event in the pathogenesis of the disease. The clinical pattern is characterized by an acute onset of the disease, with loss of vision due to macular involvement. Numerous or isolated dark lesions with perifocal, whitish-yellow zones appear in the macular area. The lesions increase in size and number and assume an appearance of cystic clusters. In the acute phase of RPE inflammation, macular changes are subtle and may be misidentified as the early phase of other retinal disorders. Even in the late phases of FA, there is hyperfluorescence at the sites of whitish-yellow zones and hypofluorescence at the dark central locations. In 6-12 weeks, reconvalescence occurs, and the visual acuity improves. In this period, the dark central lesion with its hypopigmented halo becomes less evident. These changes will remain evident for 6-7 years. Patients with acute retinal pigment epitheliitis have dark lesions appearing as cystic clusters surrounded by hypopigmented halolike zones, whereas patients with APMPPE exhibit creamy-grey and pink-grey lesions [28, 29]. Acute macular neuroretinopathy was first described in 1975 by Bos and Deutman. Its etiology and pathogenesis have not been elucidated. The disease has been associated with numerous risk factors including oral contraceptive use, allergic immune reactions, and viral illness. The condition is characterized by minor vision loss and acute-onset, paracentral scotoma. Major ophthalmoscopic findings are irregularly-shaped reddish-brown parafoveal retinal lesions which are better seen using red-free light. In several cases that presented early, features suggesting edema were present and which preceded the reddish-brown lesions. These lesions appear to lie at the level of the outer retinal layers. No signs of inflammatory reactions such as vasculitis or cellular response in the vitreous have been observed. FA shows a faint hypofluorescence corresponding with the lesions and slight dilation of retinal capillaries. An interesting aspect of the disease is the long course with continued gradual improvement in the field defect and fading of the retinal lesion. Funduscopic examination demonstrates multiple large, flat, yellow-white placoid lesions at the level of the RPE. The lesions have well-defined borders and are frequently confluent. Suspended inflammatory cells and floaters in the form of dissociated fibrils can be seen in the vitreous. Early phase FA in the disease shows marked hypofluorescence with areas of blocked fluorescence characteristic of acute lesions. The size of hypofluorescence may exceed the size of the lesions found by ophthalmoscopy in the fundus. Fluorescein angiography shows marked choroidal hypofluorescence due to blockage of fluorescence. In addition, early phase FA in the disease may show small hypofluorescent foci that cannot be detected clinically (which supposes abnormal choriocapillaris perfusion) and become hyperfluorescent in the mid and late phases. They have indistinct borders, and angiograms show large choroidal vessels crossing placoid lesions. New lesions appear over the first weeks of the disease; they are dark and hypofluorescent, whereas old lesions are hyperfluorescent and have distinct borders. As scarring progresses, there appear large zones of changed background fluorescence associated with RPE alterations, and signs of choriocapillaris occlusion disappear. Scarring occurs 2-4 weeks after the onset of the disease, and is accompanied by the formation of atrophic areas with insignificant pigment redistribution. The disease may be recurrent, leading to an increase in the number of atrophic lesions that may be confluent [28, 29]. The disease has a longer course than APMPPE. In addition, in this disease (but not in APMPPE), the lesions are located at the level of the RPE and may be confluent, and there is no tendency to vasculitis. Acute syphilitic posterior placoid chorioretinitis (ASPPC) is more frequently observed in the secondary and tertiary stages of syphilis. The disease is most common in males older than 50 years. Human Immunodeficiency Virus (HIV) infection is a frequent co-infection among patients with syphilis. A singular or multiple yellow-colored, circular, deep retinal or choroidal plaques located in the macula are characteristically present. Bilaterality occurs in about half of affected patients. The OCT findings may include focal loss of the external limiting membrane and choroidal infiltration. FA shows early hypofluorescence with a leopard spot pattern of hypo- and hyperfluorescence in the faded part of the lesions with late retinal pigment epithelial staining [28], which is not typical for APMPPE. The differential diagnosis should include serpiginous choroiditis. The latter is slightly more common in men between 30 and 70 years of age, leaves atrophic scars, and is characterized by frequent recurrences and poor functional prognosis. The disease can be classified on the basis of clinical presentation as peripapillary, macular and ampiginous. Lesions most commonly start in the peripapillary region. Active lesions are yellow to grayish with associated overlying retinal edema. Active lesions become atrophic in weeks to months, with atrophy of the retinal pigment epithelium, choriocapillaris, and choroid. On OCT, the characteristic active lesion of serpiginous choroiditis shows hyper-reflectivity and thickening of the outer retina, and increased reflectance of the choroid. FA of the active lesions shows early hypofluorescence and late hyperfluorescence in a typical geographic pattern. Retinal vessels may stain adjacent to the active lesions. In APMPPE, fluorescein angiography shows irregularly shaped non-fluorescent zones with well-defined borders, which begin to exhibit faint fluorescence a few minutes after the dye injection [28, 29]. The key difference is the clinical course. Unlike APMPPE, serpiginous choroiditis is a progressive, chronic disease, with its lesions being greyish yellow to black and located juxtapapillary. In acute Vogt–Koyanagi–Harada syndrome, the lesions develop at the level of the RPE, along with serous retinal detachment, papillary edema, inflammatory reaction in the vitreous and granulomatous inflammation in the anterior segment. In fluorescein angiography, the active process with the presence of subretinal exudation is seen as numerous hyperflourescent dots at the level of the RPE. The foci gradually increase in size and become confluent as they increase in number. In the chronic phase, angiography reveals diffusely scattered dots of hyperfluorescence due to the window defects at the level of the RPE. In APMPPE, on completion of the exudation phase, these changes are not visualized. In the acute phase, the lesions block choroidal fluorescence. In the resolution period, numerous patches of hyperfluorescence can be noted which appeared in the preretinal arterial phase and early arterial phase. In Vogt–Koyanagi–Harada syndrome, OCT reveals serous retinal detachments at the macula. The subretinal fluid may reveal a higher optical density than the vitreous, suggesting a higher level of protein content. Vitreoretinal interface alterations with cellular deposits may be seen. Subretinal fibrinoid deposits are seen, which may later evolve into subretinal fibrosis. Enhanced depth imaging and swept source OCT will show significant choroidal thickening, which resolves when treated [28-30]. Unlike APMPPE, Vogt–Koyanagi–Harada syndrome is characterized by exudative retinal detachment, macular and papillary edema and confluent lesions. In addition, patients with Vogt–Koyanagi–Harada syndrome may develop secondary glaucoma.
Conclusion Our patient was found to have an acute-onset recurrent bilateral APMPPE. OCT found numerous flat yellowish-white, grey, creamy and pink lesions in the macula and posterior pole at the level of the RPE and choriocapillary layers. Early fluorescein angiography of both eyes showed hypofluorescent lesions. The degree of hypofluorescence gradually increased with time, with the formation of areas of RPE atrophy with mildly redistributed pigment, and lesions of various severities were observed in the fundus. In the late phase of FFA, brighter fluorescence was seen in the areas of serous detachment and hyperfluorescence of the optic disc was noted, which was consistent with the classical picture of the disease. The visual acuity improved and perilesional edema substantially subsided with the treatment. At three-month follow-up, there were further improvements in visual acuity and the state of the retina and vessels, and most lesions became flatter. Although acute posterior multifocal placoid pigment epitheliopathy is a rare disease, it must be borne in mind that the differential diagnostic assessment is essential for early and adequate treatment. These patients should be followed since recurrences can occur.
References 1.Testi I, Vermeirsch S, Pavesio C. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE). J Ophthalmic Inflamm Infect. 2021 Nov 1;11(1):31. 2.Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968 Aug;80(2):177–85. 3.Jones NP. Acute posterior multifocal placoid pigment epitheliopathy. Br J Ophthalmol. 1995 Apr;79(4):384-9. 4.Saleh M. Placoid pigment epitheliopathy and serpiginous choroiditis. J Fr Ophtalmol. 2020 Feb;43(2):e55-e66. 5.Deutman AF, Lion F. Choriocapillaris nonperfusion in acute multifocal placoid pigment epitheliopathy. Am J Ophthalmol. 1977 Nov;84(5):652-7. 6.Thomson SP, Roxburgh ST. Acute posterior multifocal placoid pigment epitheliopathy associated with adenovirus infection. Eye (Lond). 2003 May;17(4):542-4. 7.Augsten R., Pfister W., Königsdörffer E. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) and borreliosis. Klin Monbl Augenheilkd. 2009;226(6):512–513. 8. Borruat FX, Piguet B, Herbort CP. Acute posterior multifocal placoid pigment epitheliopathy following mumps. Ocul Immunol Inflamm. 1998 Sep;6(3):189-93. 9.Hsu CT, Harlan JB, Goldberg MF, Dunn JP. Acute posterior multifocal placoid pigment epitheliopathy associated with a systemic necrotizing vasculitis. Retina. 2003 Feb;23(1):64-8. 10.Van Buskirk EM, Lessell S, Friedman E. Pigmentary epitheliopathy and erythema nodosum. Arch Ophthalmol. 1971 Mar;85(3):369-72. 11.Vasseneix C, Aouidid S, Brasseur G, Quintyn JC. [Multifocal placoid pigment epitheliopathy as a manifestation of ophthalmological sarcoidosis]. J Fr Ophtalmol. 2007 May;30(5):e12. French. 12.Wolf MD, Folk JC, Panknen CA, Goeken NE. HLA-B7 and HLA-DR2 antigens and acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1990 May;108(5):698-700. 13.Kraemer LS, Montgomery JR, Baker KM, Colyer MH. Acute posterior multifocal placoid pigment epiteliopathy after immunization with multiple vaccines. Retin Cases Brief Rep. 2022 Jan 1;16(1):16-19. 14.Brézin AP, Massin-Korobelnik P, Boudin M, et al. Acute posterior multifocal placoid pigment epitheliopathy after hepatitis B vaccine. Arch Ophthalmol. 1995. 1995 Mar;113(3):297-300. 15.Mendrinos E., Baglivo E. Acute posterior multifocal placoid pigment epitheliopathy following influenza vaccination. Eye. 2010;24(1):180–181. 16.O'Halloran HS, Berger JR, Lee WB, et al. Acute multifocal placoid pigment epitheliopathy and central nervous system involvement: nine new cases and a review of the literature. Ophthalmology. 2001 May;108(5):861-8. 17.Spaide RF, Yannuzzi LA, Slakter J. Choroidal vasculitis in acute posterior multifocal placoid pigment epitheliopathy. Br J Ophthalmol. 1991 Nov; 75(11): 685–7. 18.Algahtani H, Alkhotani A, Shirah B. Neurological Manifestations of Acute Posterior Multifocal Placoid Pigment Epitheliopathy. J Clin Neurol. 2016 Oct;12(4):460-467. 19.Browne AW, Ansari W, Hu M, et al. Quantitative Analysis of Ellipsoid Zone in Acute Posterior Multifocal Placoid Pigment Epitheliopathy. J Vitreoretin Dis. 2020 Jun 1;4(3):192-201. 20.Raven ML, Ringeisen AL, Yonekawa Y, Stem MS, Faia LJ, Gottlieb JL. Multimodal imaging and anatomic classification of the white dot syndromes. Int J Retina Vitreous. 2017 Mar 20;3:12. 21.Goldenberg D, Habot-Wilner Z, Loewenstein A, Goldstein M. Spectral domain optical coherence tomography classification of acute posterior multifocal placoid pigment epitheliopathy. Retina. 2012 Jul;32(7):1403-10. 22.Lee GE, Lee BW, Rao NA, Fawzi AA. Spectral domain optical coherence tomography and autofluorescence in a case of acute posterior multifocal placoid pigment epitheliopathy mimicking Vogt-Koyanagi-Harada disease: case report and review of literature. Ocul Immunol Inflamm. 2011;19(1):42–47. 23.Heiferman MJ, Rahmani S, Jampol LM, Nesper PL, Skondra D, Kim LA, Fawzi AA. Acute posterior multifocal placoid pigment epitheliopathy on optical coherence tomography angiography. Retina. 2017;37(11):2084–2094. 24.Kirkham TH, Ffytche TJ, Sanders MD. Placoid pigment epitheliopathy with retinal vasculitis and papillitis. Br J Ophthalmol. 1972 Dec;56(12):875-80. 25.Kawia AA, Wanga DZ, Kamal K, Kattaha JC. Cerebrovasc Dis. Kawi AA, Wang DZ, Kamal K, Kattah JC. A case of ischemic cerebral infarction associated with acute posterior multifocal placoid pigment epitheliopathy, CNS vasculitis, vitamin B(12) deficiency and homocysteinemia. Cerebrovasc Dis. 2004;18(4):338-9. 26.Hammer ME, Grizzard WS, Travies D. Death associated with acute, multifocal, placoid pigment epitheliopathy. Arch Ophthalmol. 1989 Feb;107(2):170-1. 27.Quillen DA, Davis JB, Gottlieb JL, et al. The white dot syndromes. Am J Ophthalmol. 2004;137(3):538–550. 28.Heimann H, Kellner U, Foerster MH. Atlas of Fundus Angiography. 1st Edition ed. Stuttgart, New York: Thieme, 2006 29.Duker JS, Waheed NK, Goldman DR. Handbook of retinal OCT. 2nd Edition. Elsevier Science; 2021. 30.Konovalova NV, Khramenko NI, Guzun OV, Slobodyanik SB, Kovtun AV, Urchenko LA. A case of Vogt-Kayanagi-Harada syndrome. Oftalmologiia Vostochnaia Evropa. 2020;10(2):260-75. Russian.
Disclosures Authour Contribution: Konovalova N V: Conceptualization, Project administration, Data Curation, Formal Analysis, Writing – original draft; Khramenko NI: Conceptualization, Investigation, Writing – review & editing; Slobodianyk SB: Data Curation, Writing – review & editing; Kovtun OV: Writing – original draft, Data Curation. Conflict of Interest Statement: The authors state that they have no conflict of interest that might bias this work.
|