J.ophthalmol.(Ukraine).2022;5:37-41.
|
http://doi.org/10.31288/oftalmolzh202253741 Received: 26.09.2022; Accepted: 11.10.2022; Published on-line: 27.10.2022 Neuroophthalmic abnormalities in supranuclear palsy L. V. Venger 1, I. V. Khubetova 2 1 Odesa National Medical University 2 Odesa regional clinical hospital Odesa (Ukraine) TO CITE THIS ARTICLE: Venger LV, Khubetova IV. Neuroophthalmic abnormalities in supranuclear palsy. J.ophthalmol.(Ukraine).2022;5:37-41. http://doi.org/10.31288/oftalmolzh202253741
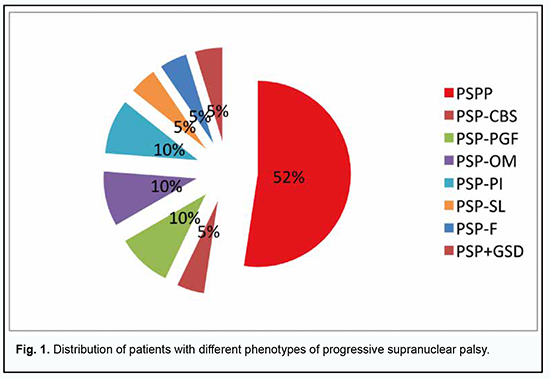
Background: Progressive supranuclear palsy (PSP) is the most common atypical parkinsonism with various movement disorders and oculomotoric abnormalities. Purpose: To identify major neuroophthalmic manifestations in different clinical phenotypes of PSP. Material and Methods: The study was conducted at the Odesa regional clinical hospital in 2011 to 2021. Twenty one patients with PSP (including one patient with PSP combined with Hallervorden–Spatz disease and levodopa-induced hyperkynesis) underwent an examination. This included a classical neurological examination and comprehensive neuroophthalmic examination with high-field magnetic resonance imaging (MRI) of the brain and spine and videonystagmography testing. Mean patient age was 53.2 ± 1.1 years, and most patients were women (13 [61.9%]). Statistical analysis included frequency analysis. Results: PSP-P was the most common phenotype of PSP (52.4%), followed by PSP-PGF (9.5%), PSP-OM (9.5%), PSP-PI (9.5%), PSP-CBS (4.8%), PSP-SL (4.8%), PSP-F (4.8%) and PSP+GSD (4.8%). All patients showed changes in velocity and amplitude of vertical saccades. Diplopia was, however, observed in only 12 (57.1%) patients. Conclusion: Neurological and neuroophthalmic examination is decisive in establishing the diagnosis of progressive supranuclear palsy and selecting a treatment strategy. In the current study, PSP-P was the most common (52.4%) phenotype, the frequency of PSP-OM variant was 9.5%. Keywords: progressive supranuclear palsy, neuroophthalmology, diagnostic assessment, phenotype
Introduction Progressive supranuclear palsy (PSP) is a 4R tauopathy and is a subtype of frontotemporal lobar degeneration with tau inclusions (FTLD-tau). Tau is a microtubule-associated protein with versatile functions in the dynamic assembly of the neuronal cytoskeleton. Four-repeat (4R-) tauopathies are a group of neurodegenerative diseases defined by cytoplasmic inclusions predominantly composed of tau protein isoforms with four microtubule-binding domains [1, 2]. The post-mortem diagnosis of PSP is dependent on the identification of neurofibrillary tangles and threads in subcortical nuclei; tufted astrocytes and coiled bodies are other highly characteristic findings [1]. In addition, autopsy studies have demonstrated coiled oligodendroglial bodies in the extracellular matrix and signs of diffuse cytoplasmic immune reactivity of neurons [1, 2]. Biochemical and pathohistological markers have been found that allow differentiating PSP with predominantly parkinsonian symptoms (PSP-P) from the classical clinical phenotype of Steele–Richardson–Olszewski syndrome (PSP-RS) [1, 3, 4]. Other PSP variants/phenotypes have been recognized. In 2017, the Movement Disorders Society put forward new clinical criteria for the diagnosis of PSP recognizing diverse PSP phenotypes including PSP-RS, PSP-P, and PSP with predominant corticobasal syndrome (PSP-CBS), with predominant progressive gait freezing (PSP-PGF), with predominant ocular motor dysfunction (PSP-OM), with predominant postural instability (PSP-PI), with predominant frontal presentation (PSP-F), and with predominant speech and language disorder (PSP-SL) [5]. The prevalence of different PNP phenotypes has been studied. In a 2007 study by Williams and colleagues [6], the mean severity in all regions of the RS group was higher than in PSP-P and PSP-PGF and the overall tau load was significantly higher in PSP-RS than in PSP-P. Sakae et al. [7] compared PSP-RS cases with PSP-F and found increased tau burden only in the superior frontal gyrus gray matter and inferior temporal gyrus white matter in PSP-F. Tsuboi and colleagues [8] investigated clinicopathological characteristics of 5 cases of PSP that presented with corticobasal syndrome (CBS-PSP). They concluded that when PSP presents as CBS, it is most likely due to either a concurrent cortical pathology from a secondary process such as Alzheimer's disease or from the primary pathology of PSP extending into cortical areas that are primarily and commonly affected in corticobasal degeneration. Ling et al. [8] also examined PSP-CBS cases and demonstrated that the overall severity of tau pathology was the same between PSP-CBS and PSP-RS but with a shift of tau burden towards the cortical regions. On the other hand, the rare PSP-PGF variant showed almost no cortical tau pathology, but severe degeneration of the globus pallidus, substantia nigra, and subthalamic nucleus, hence called also pallido-nigro-luysian degeneration [8]. In addition to the recognition of clinical subtypes, a novel concept raises the possibility of propagation of pathological tau in PSP as well as other tauopathies [5] providing a potential therapeutic target [5, 9]. Indeed, sequential distribution patterns have been recognized for tau pathologies such as neurofibrillary degeneration in Alzheimer’s disease (AD) [1, 10], Pick’s disease [11], argyrophilic grain disease [12], or astrocytic tau pathologies [13] as well as for other proteinopathies such as beta-amyloid, alpha-synuclein or TDP-43. Regarding PSP, the clinical manifestations (and, consequently, the phenotype) of the disease depend on the brain compartments in which tau protein accumulates, and on the concentration of tau protein. Thus, in low involvement of the globus pallidus, striatum and substantia nigra, with rare involvement of the premotor cortex, tau protein accumulates at low concentrations or does not accumulate in the regions (such as the dentate nucleus, cerebellar white matter and pons) that are more caudal than the substantia nigra, and clinical manifestations are usually absent. In these episodes, tau protein does not accumulate in the parietal cortex and accumulates at very low concentrations in the anterior frontal lobe. The disease is most commonly found when there is the central involvement of the basal ganglia and dentate nucleus with frontal and parietal lesions. The inner globus pallidus, subthalamic nucleus, substantia nigra, pontine nucleus, dentate nucleus and cerebellar white matter are affected more severely, but with no increase in the grade of coiled body plus thread lesions in the outer globus pallidus. The cortical regions including the frontal and parietal cortex are affected moderately. Neuroophthalmic manifestations in PSP are of special interest. Early neuroophthalmic manifestations of PSP are slow vertical saccades and square-wave jerks (SWJ), small amplitude (0.5-5 degrees) refixational eye movements [1, 14, 15]. Supranuclear ophthalmoplegia (classic gaze palsy in PSP) is caused by lesions of the supranuclear pathways [16], with the nuclei of cranial nerves, neural bundles, neuromuscular synapses and extraocular muscles remaining intact. Improvement in supranuclear vertical gaze limitation after extravolitional pathway activation with the vestibular ocular reflex (VOR) or the Bell phenomenon has been reported [14]. The Bell phenomenon consists of a reflex upward movement of the eye globe with eye lid closure. The Bell’s phenomenon can be assessed by the examiner elevating the patient’s upper eyelids using his finger and thumb of one hand, asking the patient to close his eyes. The vertical VOR can be activated by manually flexing and extending the neck while the patient views a distant target. If the extent of the vertical eye movement limitation is improved with either of these maneuvers, then the lesion is supranuclear in origin [14]. Despite advances in studies of visual abnormalities in patients with PSP, the courses of the clinical variants of the disease have been not sufficiently investigated, which warrants further analysis using a multidisciplinary approach. The purpose of the study was to identify major neuroophthalmic manifestations in different clinical phenotypes of PSP. Material and Methods The study was conducted at the Odesa regional clinical hospital in 2011 to 2021. Twenty one patients with PSP (including one patient with PSP combined with Hallervorden–Spatz disease and levodopa-induced hyperkynesis) underwent an examination. This sample constitutes 3.7% of the registered patients with extrapyramidal pathology in the region; this is in agreement with the data reported by others. In addition to a classical neurological examination, patients underwent a neuroophthalmic examination which included visual acuity, contrast sensitivity, color vision, slit-lamp biomicroscopy and ophthalmoscopy of the anterior and posterior segments, automatic visual field testing (Tomey AP-3000, Nagoya, Japan) and optical coherence tomography (OCT). Moreover, they underwent high-field magnetic resonance imaging (MRI) of the brain and spine, and videonystagmography testing using video Frenzel system (Vestibular, Russian Federation). Mean patient age was 53.2 ± 1.1 years, and most patients were women (13 [61.9%]). Statistical analysis included frequency analysis. Measures of central tendency, dispersion and percentiles were calculated. Results The course of the disease was typical in all patients. Major complaints included gait instability and abnormal movements with falls, changes in behavior, impatience or apathy, muscle rigidity, inability to control movements of the eyes and lids (e.g., focusing on particular objects, upgaze and downgaze), and slow, soft speech. The onset of the disease was gradual and patients noted fatigue, frequent headaches, dizziness and arthralgia in early disease. Half of patients experienced depression. Mild changes in personality were seen in 66.7% of patients, and memory problems, in 81.0% of patients. Pseudobulbar symptoms were observed in most patients (16 [76.2%]). All patients exhibited manifestations of supranuclear ophthalmoplegia and cervical dystonia. Marked parkinsonism symptoms with frequent falls over and abnormal postural reflexes and minimum response to treatment with levodopa were noted in 12 (57.1%) patients. Bradykinesia, amimia and frightened face were typical features. One (4.8%) patient exhibited marked behavioral and cognitive abnormalities typical for front brain lesions. In another patient, the disease was manifested by marked abnormality of gait and static and dynamic functions in general. The neurological status of patients was estimated. Nineteen (90.5%) patients had a postural deficit (a positive pull test). Axial rigidity was the most common phenomenon (85.7%). Dysarthria and monotonous speech with mild hypophonia were common. Neuroimaging found the “hummingbird” sign (a midbrain to pons ratio ≤ 0.16) in 17 (81.08%) patients. PSP-P was the most common phenotype of PSP (52.4%), followed by PSP-PGF (9.5%), PSP-OM (9.5%), PSP-PI (9.5%), PSP-CBS (4.8%), PSP-SL (4.8%), PSP-F (4.8%) and PSP+GSD (4.8%) (Fig. 1).
All patients showed changes in velocity and amplitude of vertical saccades, with the greatest changes seen in patients with PSP-OM. Diplopia was, however, observed in only 12 (57.1%) patients. Since the diagnosis of PSP can be confirmed only post mortem, the correct use of lifetime diagnostic criteria for particular PSP variants enables not only establishing the diagnosis, but also determining the prevalent phenotype with high probability. An assessment of the alignment of the eyes in extreme gaze positions at near and at distance frequently determines the origin of symptoms of diplopia. An assessment of the eyelid state and motility may also supply important information. Typical personality traits, especially those associated with dysarthria, may produce a clinical picture close to the pathognomonic picture. Studying rotatory and tracking movements of the eye is also important. Although neuroophthalmic symptoms of PSP are usually detected early, they may be infrequently absent at the onset of the disease. PSP cases with no neuroophthalmic symptoms have been rarely reported. Slow vertical saccades and quick phases are often the earliest symptom of PSP. Classic vertical supranuclear ophthalmoparesis appears later, with downgaze affected more than upgaze. In PSP-RS, vertical movements of the eyes were triggered by VOR until the late stages of the disease, although the Bell’s phenomenon (a reflex upward movement of the eye globe with eye lid closure) was often absent. In the later stages of the disease, ophthalmoparesis affects both horizontal and vertical eye movements, with a reduction in the amplitude of these movements. Total ophthalmoparesis may develop in the terminal stages of the disease. Almost continuous saccadic oscillations during periods of fixation are observed in all patients with PSP. These are horizontal movements with a small amplitude of 5° that disrupt fixation and, after a 180-200-ms delay, bring the eyes back to the target. Unlike occasional saccadic oscillations, longer saccadic oscillations are often seen in elderly individuals with PSP. Abnormal convergence was noted in patients experiencing occasional diplopia at near. In 2 (9.5%) patients, diplopia was associated with decompensated orthophoria secondary to abnormal binocular fusion. Abnormal VOR suppression was found in a patient with PSP-OM in the presence of diplopia. Eyelid symptoms (lid retraction, apraxia of eyelid opening or closing, blepharospasm or Graefe’s symptom) were seen in 16 (76.2%) patients with PSP. Loss of the fast component of the optokinetic nystagmus preceded gaze palsy. Pupil abnormalities included a reduced pupil diameter in darkness compared with controls. Below we describe an example case of PSP. A female born in 1967 was admitted to the hospital in December 2020, and reported deterioration over the previous three years in her health with postural changes, neck and back pain, and a deterioration of memory. Her gait worsened, she began exhibiting a frozen gaze, and her face changed over a month before admission. The patient was hospitalized to the neurology ward for examination for suspected Steele–Richardson–Olszewski disease due to increasing motor abnormalities (changes in gait, rigid muscles, mostly in the neck and shoulder region). A neurological examination showed asthenic habitus and hypomimia. Palpebral fissures were equal and pupils equal, round and reactive to light. Sometimes her gaze appeared frozen. There were slow horizontal saccades, slight upward gaze paresis, positive Bell’s phenomenon, and no change in facial sensitivity. The patient was able to extend her tongue along the midline and had no bulbar symptoms. Tendon and periosteal reflexes were equal and normal in all limbs. No pathological reflexes were elicited. Neck and upper limb muscles were rigid. The cogwheel symptom as well as digital rigidity test was positive. The patient had a positive pull test. When walking, the patient experienced axial rigidity (rigid neck and shoulder muscles) and bilateral acheirokinesis. In addition, when turning her body, she was turning her trunk and neck simultaneously. Her Mini Mental State Examination (MMSE) score was 26. MRI scanning showed no evidence of pontine changes, and the anteroposterior length of the midbrain was as short as 1.2 cm. In addition, the midbrain to pons ratio (MPR) was 0.6, which corresponded to the average population value. Therefore, a neurological and neuroophthalmic examination was decisive in establishing the diagnosis of PSP and selecting a treatment strategy in this patient. Discussion Internuclear ophthalmoplegia is an uncommon neuroophthalmic manifestation, but lesions of the medial longitudinal fasciculus (MLF) are common neuroophthalmic manifestations in PSP, but not in acute cerebrovascular accidents [16]. Steele and colleagues [17] in their original report on the neuropathological findings of PSP, noted the possibility of bilateral MLF lesions in PSP. Mastaglia and Grainger [18, 19] reported patients clinically diagnosed as having PSP who presented with oculomotor abnormalities including bilateral MLF syndrome. Others also reported on bilateral MLF lesions in patients clinically diagnosed as having PSP [6, 10]. Wall-eyed bilateral internuclear ophthalmoplegia (WEBINO) syndrome is manifested by primary position exotropia and bilateral divergence, develops in bilateral rostral lesions of the MLF and has been reported in patients with PSP [3]. This variant of ophthalmoplegia was not found in patients of the current study. Irrespective of the prevailing clinical variant, the most common neuroophthalmic manifestations in PSP are slowed vertical saccades and slowed quick phases followed by saccadic oscillations, with the greatest changes seen in patients with PSP-OM. Because there is marked variability in the clinical picture of PSP and the disease has several phenotypes, early comprehensive neuroophthalmic examination is especially important for adequate lifetime diagnostic assessment. Conclusion First, neurological and neuroophthalmic examination is decisive in establishing the diagnosis of progressive supranuclear palsy and selecting a treatment strategy. Second, PSP-P was the most common (52.4%) and PSP-OM was a less common (9.5%) PSP variant in the current study. Finally, in the current study, the major neuroophthalmic manifestations of PSP were slowed vertical saccades and slowed quick phases followed by saccadic oscillations.
References 1.Tagai K, Ono M, Kubota M, Kitamura S et al. High-Contrast in Vivo Imaging of Tau Pathologies in Alzheimer’s and Non-Alzheimer’s Disease Tauopathies. Neuron. 2021 Jan 6;109(1):42-58.e8. 2.Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci. 2011 Nov;45(3):384-9. 3.Robinson JL, Yan N, Caswell C, Xie SX, Suh E, Van Deerlin VM et al. Primary Tau Pathology, Not Copathology, Correlates With Clinical Symptoms in PSP and CBD. J Neuropathol Exp Neurol. 2020 Mar 1;79(3):296-304. 4.Levin J, Kurz A, Arzberger T, Giese A, Höglinger GU. The Differential Diagnosis and Treatment of Atypical Parkinsonism. Dtsch Arztebl Int. 2016 Feb 5;113(5):61-9. 5.De Pablo-Fernández E, Lees AJ, Holton JL, Warner TT. Prognosis and Neuropathologic Correlation of Clinical Subtypes of Parkinson Disease. JAMA Neurol. 2019 Apr 1;76(4):470-479. 6.Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, Revesz T. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain. 2007 Jun;130(Pt 6):1566-76. 7.Sakae N, Josephs KA, Litvan I, Murray ME, Duara R, Uitti RJ, Wszolek ZK, Graff-Radford NR, Dickson DW. Neuropathologic basis of frontotemporal dementia in progressive supranuclear palsy. Mov Disord. 2019 Nov;34(11):1655-1662. 8.Tsuboi Y, Josephs KA, Boeve BF, Litvan I, Caselli RJ, Caviness JN et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord. 2005 Aug;20(8):982-8. 9.Przewodowska D, Marzec W, Madetko N. Novel Therapies for Parkinsonian Syndromes-Recent Progress and Future Perspectives. Front Mol Neurosci. 2021 Aug 26;14:720220. 10.Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017 Jul;16(7):552-563. 11.Kovacs GG, Lukic MJ, Irwin DJ, Arzberger T, Respondek G, Lee EB et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020 Aug;140(2):99-119. 12.Choi JH, Kim H, Shin JH, Lee JY, Kim HJ, Kim JM, Jeon B. Eye movements and association with regional brain atrophy in clinical subtypes of progressive supranuclear palsy. J Neurol. 2021 Mar;268(3):967-977. 13.Tolnay M, Clavaguera F. Argyrophilic grain disease: a late-onset dementia with distinctive features among tauopathies. Neuropathology. 2004 Dec;24(4):269-83. 14.Narasimhan S, Changolkar L, Riddle DM, Kats A, Stieber A, Weitzman SA et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J Exp Med. 2020 Feb 3;217(2):e20190783. 15.Friedman DI, Jankovic J, McCrary JA. Neuro-ophthalmic Findings in Progressive Supranuclear Palsy. J Clin Neuroophthalmol. 1992 June; 12 (2): 104-109. 16.Progressive Supranuclear Palsy https://eyewiki.aao.org/Progressive_Supranuclear_Palsy. 17.Muratova TM, Venger LV, Khramtsov DM, Vorokhta IuM, Teliushchenko VD. Neuro-ophthalmological abnormalities in patients with ischemic stroke in the setting of a stroke center of a university clinic. J Ophthalmol (Ukraine). 2020;5:56-61. 18.Agarwal S, Gilbert R. Progressive Supranuclear Palsy. 2021 Apr 11. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan. 19.Mastaglia FL, Grainger KM. Internuclear ophthalmoplegia in progressive supranuclear palsy. J Neurol Sci. 1975 Jul;25(3):303-8.
Disclosures Author Contributions: Venger LV: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing Khubetova IV: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing – original draft All authors reviewed the results and approved the final version of the manuscript. Funding sources: No stated funding sources. Conflict of interest: The authors state that they have no conflict of interest that might bias this work.
The study involved human subjects, was approved by the Ethics Committee, and adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from all participants. This study did not include animal experiments.
|