J.ophthalmol.(Ukraine).2020;6:64-69.
|
http://doi.org/10.31288/oftalmolzh202066469 Received: 05 June 2020; Published on-line: 21 December 2020 ANA-associated uveitis in the presence of reactivated HHV-7 infection in a patient with MBL deficiency D. V. Maltsev1, O. O. Hurzhii2 1 Institute of Experimental and Clinical Medicine, Bogomolets National Medical University; Kyiv (Ukraine) 2 Ailas Clinic; Kyiv (Ukraine) TO CITE THIS ARTICLE: Maltsev DV, Hurzhii OO. ANA-associated uveitis in the presence of reactivated HHV-7 infection in a patient with MBL deficiency. J.ophthalmol.(Ukraine).2020;6:64-9. http://doi.org/10.31288/oftalmolzh202066469 Bilateral, ANA-positive uveitis developed in a patient with primary total mannose binding lectin (MBL) deficiency during human herpes virus type 7 (HHV-7) reactivation from latently infected salivary glands. The diagnosis of uveitis was confirmed based on eye examination, findings of optical coherence tomography, and elevated serum antinuclear antibody titer (1:320). HHV-7 reactivation from persistence was verified by blood white cell polymerase chain reaction (PCR) and eye swab PCR. The patient was diagnosed with primary MBL deficiency based on the results of enzyme-linked immunosorbent assay and special genetic testing. The serum MBL level was zero, but all other studied characteristics were within respective reference ranges. We identified three pathologic polymorphisms (223 С/Т, 230 G/A, and 239 A/G) in the MBL-2 gene, which indicated a genetic origin for the detected immunodeficiency. The results of these tests provided a rationale for administering a comprehensive treatment for suppression of the autoimmune process (rituximab and methotrexate), elimination of HHV-7 DNA from blood white cells and ocular smears (valganciclovir) and compensation of primary immunodeficiency by replacement therapy with intravenous infusion of solvent-detergent-treated fresh frozen plasma containing MBL. Our comprehensive approach to treatment led not only to a complete remission of uveitis, but also to compensation of other autoimmune, allergic and infectious symptoms and signs of the immunodeficiency. The data from this report expand our knowledge on the heterogeneity of clinical signs and symptoms of primary MBL deficiency in humans and provide pathways to studies on new potential pathogenetic mechanisms of autoimmune lesions in this genetic immune dysfunction which is one of the most common in the population. Keywords: uveitis, HHV-7, mannose binding lectin deficiency
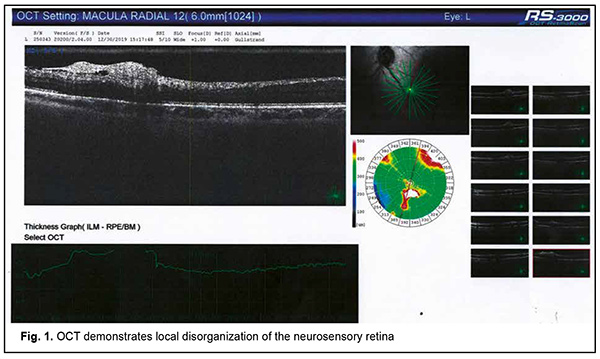
Autoimmune uveitis is a severe progressive immune inflammatory ocular disease, which can lead to rapid loss of visual function if not treated promptly [1]. Recently, advances have been made in the treatment and our understanding of the mechanisms of the disease. It has been found that the disease may be induced by microbial triggers under conditions of immunosuppression. Herpes viruses occupy a special place among the microbial triggers that can promote the breakdown of immune tolerance with the development of autoimmune uveitis [2]. Since these opportunistic agents are re-activated in the body of an immunocompromised individual through decreased immune surveillance, assessing immune status for finding the causative immune deficiency is an important component of rational diagnostics in such cases [3, 4]. It should be taken into account that it is not only secondary immune deficiencies, but also primary immune deficiency diseases, including genetically determined minor immune deficiencies, that can cause herpes virus reactivation from persistence in biological reservoirs [5]. Therefore, it is reasonable to use a three-component approach to a diagnosis of autoimmune uveitis: (1) verification of uveitis itself, (2) identification of the trigger which provoked an autoimmune response to ocular autoantigens, and (3) the diagnosis of causative immune deficiency which contributed to reactivation of the trigger and caused alterations in the mechanisms of maintenance of immune tolerance in the human body. This may ensure development of a comprehensive approach to the treatment of autoimmune uveitis with simultaneous systemic effects on all the main components of disease pathogenesis, thus not only suppressing the autoimmune response against ocular autoantigens, but also removing the reactivated microbial trigger and compensating for the immunodeficiency disease which has mediated the development of ocular lesions through decreased immune surveillance for the trigger and support of immune tolerance to host antigens [6]. Here we report a case demonstrating our multidisciplinary triple-component approach to the diagnosis of patients with autoimmune uveitis, which takes into account the main components of disease pathogenesis, thus enabling the development of an effective comprehensive approach to treatment. A female patient, born in 1967, presented to the ophthalmologist with complaints of progressive bilateral loss of vision and transient ocular pain. It was known from her history that she had been ill for the last 5 years. The disease had a waxing and waning course with short periods of remissions changed by exacerbations. The patient was diagnosed with autoimmune uveitis of the right eye in early disease and with autoimmune uveitis of the left eye, two years later. By the time of presentation, she had undergone three courses of plasmapheresis and several courses of systemic and topical corticosteroid therapy, with some transient improvements in her condition. In addition, she had undergone an episode of macular edema of the right eye. Moreover, in the presence of hormonal therapy, she had several times developed severe conjunctival blepharitis, which required administration of antibiotics in addition to glucocorticosteroids. At presentation, her best-corrected visual acuity (BCVA) was 0.4 in the right eye and 0.6 in the left eye. She exhibited bilateral signs of early corneal and lens opacification, with decreased transparency of the vitreous and fibrous vitreous strands in both eyes. Optical coherence tomography of the left eye demonstrated focal changes in the paramacular retinal area (Fig. 1, panel 3). A focus of disorganized inner retinal layer was seen in a series of scans in the region of the inferotemporal vascular arcade. The contour, structure and differentiation of the inner neuroretinal layers appeared altered. A dense and partially disrupted vitreoretinal interface was seen.
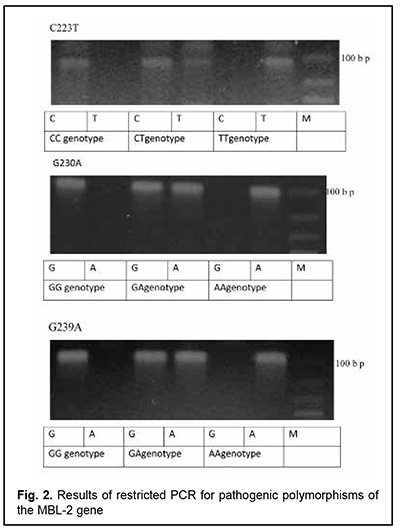
The patient had autoimmune thyroiditis caused by thyroperoxidase antibodies (she was euthyroid in the presence of L-thyroxine replacement therapy) and allergic syndrome with recurrent urticaria and angioedema. She had a history of an episode of angioedema developed as a response to a topical parabulbar dexamethasone injection for an exacerbation of uveitis. Throughout her previous life, she had had recurrent sinopulmonary bacterial infections, for which she had been treated numerous times with antibiotics and several times, on an in-patient basis, due to severe clinical condition. In addition, she had a history of recurrent urogenital bacterial infections, and, for this reason, had been registered for regular medical check-ups at a local gynecologist. For all these reasons, she was referred to the clinical immunologist for an immunological laboratory examination and assessment of her immune status. The clinical immunologist had at least 3 main tasks: (1) to confirm or deny the autoimmune nature of uveitis, since it was not known definitely whether this was done previously or not; (2) to identify the trigger of autoimmunity, and (3) to diagnose the causative immunodeficiency that could explain the development of the patient’s immunodependent syndromes (the autoimmune syndrome (uveitis, thyroiditis), allergic syndrome (urticaria, angioedema, and medication allergy) and infectious (persistent recurrent bacterial sinopulmonary and urogenital infections)) that on the first glance seemed to be separate ones, and to unify them into an integral whole. Laboratory evaluation was performed to verify the nature of uveitis, and included serum rheumatoid factor, antinuclear antibodies, anti-cyclic citrullinated peptide (ACCP) antibodies, antibodies against double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA), antibodies to neutrophil cytoplasm antigens, antimitochondrial antibodies (AMAs) and antiscleroderma antibodies and myositis blot. Blood white cell PCR and eye swab PCR with specific primers for HSV1/2, VZV, EBV, CMV, HHV-6, HHV-7, HHV-8, adenoviruses, enteroviruses, TTV, B19 parvovirus, hepatitis В, С, D and G, T. gondii, Borrelia burg., Chlamydia pneum., and Mуcoplasma pneum were performed at the Neurobiochemistry Laboratory of the Romodanov Neurosurgery Institute to find microbial triggers for the autoimmune process. This list was formed as per the data from a systematic review by Krichevskaia and Katargina [7] which discusses viral and non-viral triggers of autoimmune uveitis in humans and mechanisms of failure of immune tolerance to ocular antigens in reactivation of viral agents, particularly, molecular mimicry, bystander activation, epitope spreading, superantigen effect and effect on intestinal microbiota. Immunological evaluation included complete blood cell count and lymphocyte subpopulation analysis by flow cytometry on a Beckman Coulter Epics XL flow cytometer (Beckman Coulter, Miami, Fla.) and indirect immunofluorescence with monoclonal antibodies to CD markers with two or three immunofluorescent labels (CD3+, CD3+CD4+, CD3+CD8+, CD3–CD19+, CD3–CD16+CD56+, CD3+CD16+CD56+) (reagents from Beckman Coulter). Phagocytosis was assessed using the latex test; phagocytic number (the number of pathogenic units engulfed by one phagocyte), phagocytic index (percentage of phagocytes involved in phagocytosis), number of active phagocytes and phagocytic capacity of blood (the number of pathogenic units that can be neutralized by the phagocytes contained in 1 liter of blood) were determined. Serum levels of IgM, IgG and IgA were determined by the radial immunodiffusion method of Mancini, whereas those of IgE, IgD, IgG1, IgG2, IgG3 and IgG4, using enzyme immunoassay (Vector Best, RF). Myeloperoxidase activity of neitrophils was assessed using enzyme immunoassay at the Neuroimmunology Laboratory of the Romodanov Neurosurgery Institute. The Nitro Blue Tetrazolium assay was performed at the Zabolotnyi Institute of Microbiology and Virology as well at the Neuroimmunology Laboratory, Romodanov Neurosurgery Institute, NAMS of Ukraine. Serum level of mannose binding lectin was determined by enzyme-linked immunosorbent assay (ELISA) in Germany with assistance of Dr. Rödger Laboratory (Kyiv, Ukraine). A battery of diagnostic laboratory tests showed an increased titer of serum antinuclear antibodies (ANA 1:320), and the autoimmune nature of uveitis was confirmed. The retest for serum antinuclear antibodies produced the same results. Therefore, we found that the patient had antinuclear antibody (ANA)-positive uveitis [8]. Blood white cell PCR and eye swab PCR identified the DNA of HHV-7 (100,000 particles per sample). Apart from HHV-7, no other microbial trigger was found. HHV-7 is an opportunistic viral agent that is reactivated from persistence in the salivary glands in the body of an immunocompromised individual and is knows as a trigger of a number of human autoimmune disorders such as autoimmune thyroid disease [9], multiple sclerosis [10, 11] and autoimmune anti-NMDA encephalitis [12]. To the best of our knowledge, there have been no reports on association of ANA-positive uveitis with a reactivated HHV-7 infection. All the studied immune status characteristics were within normal range or increased, with the exception of serum mannose binding lectin (MBL) level: the patient was found to have complete MBL deficiency. The retest for serum MBL produced the same results. We performed tests for pathologic polymorphic variations within the promoter region and within the region of MBL-2 to find the cause of complete MBL deficiency in the patient, and identified three pathologic polymorphisms (223 С/Т, 230 G/A, and 239 A/G) in the gene (Fig. 2). Therefore, a clinical diagnosis of primary MBL deficiency was confirmed for the patient.
MBL deficiency is a common primary immunodeficiency which affects the lectin activation pathway of complement, is found in 5-10% of the population, and places a substantial health burden on affected individuals [5]. This immunodeficiency is reflected in liability to various infectious, autoimmune, allergic, immunoinflammatory, and oncological syndromes and some additional manifestations with a complex pathogenesis [13]. Because studies have reported on associations of MBL deficiency with severe infections, herpes simplex virus (HSV)-1 [3] and -2 [4], Epstein-Barr virus [14], cytomegalovirus [15], HSV-6 and -7 [16], papillomaviruses [17] and ТТ virus [18], our finding of reactivation of HHV-7 under conditions of this immunodeficiency in the patient can be considered natural. There have been reports on associations of MBL deficiency with a number of human autoimmune diseases such as systemic lupus erythematosus [19], rheumatoid arthritis [20], Behçet's disease [21], autoimmune spondyloarthropathy [22], autoimmune thyroid disease [23] and rheumatic fever [24]. To the best of our knowledge, this is the first report on the association of MBL deficiency with ANA-positive uveitis. Studies have reported on associations of autoimmune uveitis with primary antibody deficiencies (PADs) and complement protein deficiencies, such as common variable immune deficiency and selective IgA deficiency [25, 26], CD8 deficiency [27] and X-linked chronic granulomatous disease [28]. In a study by Kubicka-Trzaska, of the 50 patients with idiopathic posterior uveitis, 11 exhibited the deficiency of serum IgG, and 34, abnormalities of the complement system [29]. Spârchez and colleagues [28] also reported on the association of primary complement and antibody deficiencies with autoimmune uveitis in humans [30]. Therefore, our patient was clinically diagnosed with primary MBL deficiency (223 С/Т, 230 G/A, 239 A/G MBL-2): the autoimmune syndrome in the form of chronic ANA-positive autoimmune uveitis and thyroiditis associated with reactivated HHV-7 infection as a trigger of the autoimmune disease; allergic syndrome (recurrent atopic urticaria, angioedema and episodes of medication allergy); and infectious syndrome (persistent recurrent bacterial sinopulmonary and urogenital infections). This diagnosis reflects the main components of the pathogenesis of the eye disease: (a) the causative immunodeficiency that contributes to autoimmunity through alterations in the mechanisms of maintenance of immune tolerance and loss of control over the microbial trigger (MBL deficiency); (b) the microbial trigger that caused failure of immune tolerance to ocular antigens e.g., through a mechanism of molecular mimicry (reactivated HHV-7 infection); and (c) clinical consequence, autoimmune uveitis (ANA 1:320) and other autoimmune, allergic and infectious manifestations of immunodeficiency. This approach (a) unifies the immunodependent syndromes (that on the first glance seemed to be separate ones) developed by the patient during ontogenesis into integral whole, and (b) mediates a systemic view on the state of her health. This enables clinicians to administer a highly effective combination treatment aimed not only on inhibiting the autoimmune process, but directed also at the microbe trigger and immunodeficiency disease, which may give a rather prompt, apparent and durable clinical effect. Our patient was given rituximab (anti-CD 20 B cell monoclonal antibody) by intravenous infusions at a dose of 500 μg/kg body weight/month, along with methotrexate 15 mg/week orally to suppress autoimmunity to ocular autoantigens [31]. In addition, to inhibit HHV-7 activity, oral therapy with valganciclovir (450 mg twice daily) was administered guided by the results of blood white cell PCR testing. MBL deficiency was treated by replacement therapy with intravenous infusion of solvent-detergent-treated fresh frozen plasma (Octaplas, Octapharma AG, Lachen, Switzerland; 10 mL/kg body weight/twice a month), guided by the results of serum MBL testing [32]. Our comprehensive approach to treatment led to a complete and durable remission of the autoimmune process in the eye as early as one month after initiation of therapy. The HHV-7 DNA was eliminated from blood white cells and ocular smears as early as two months after initiation of continuous antiviral treatment [32]. The patient achieved the minimum normal level of serum MBL (450 ng/mL) after two infusions of plasma. Our comprehensive approach to treatment led not only to the suppression of symptoms of uveitis, but also to improvements in autoimmune thyroiditis (the patient remained euthyroid after withdrawal of the replacement therapy with L-thyroxin), allergic syndrome (no allergic events were noted throughout the 6-month follow-up) and infectious syndrome (no bacterial infection events in the upper respiratory tract or urogetinal tract were recorded in spite of the administration of immunosuppressors). The data from this report expand our knowledge on the heterogeneity of clinical signs and symptoms of primary MBL deficiency in humans and provide pathways to studies on new potential pathogenetic mechanisms of autoimmune lesions in this common genetic immune dysfunction. We have demonstrated the advantages of a multidisciplinary approach to examination and treatment of a patient with autoimmune uveitis, with involvement of the ophthalmologist, rheumatologist, clinical immunologist and infectiologist, and establishment of a multidisciplinary working group. This enables making a complete diagnosis that encompasses the complete clinical phenotype of the disease, with identification of the causative immunodeficiency, potential autoimmunity trigger and type of ocular lesions. In addition, this enables administration of comprehensive, advanced therapy targeting the main pathogenetic components of disease. We have already demonstrated advantages of the above multidisciplinary approach and comprehensive therapy in our report of a case of Toxoplasma chorioretinitis in primary myeloperoxidase MBL deficiency [6]. An advanced understanding of the problems of autoimmune diseases in humans requires an in-depth approach to making a diagnosis and conducting therapeutic treatment, which will significantly improve early and late treatment outcomes. References 1.Sève P, Kodjikian L, Adélaïde L, Jamilloux Y. Uveitis in adults: What do rheumatologists need to know? Joint Bone Spine. 2015 Oct;82(5):308–14. 2.Pleyer U, Winterhalter S. Diagnostic and therapeutic aspects of herpes virus associated uveitis. Klin Monbl Augenheilkd. 2010 May;227(5):407–12. doi: 10.1055/s-0029-1245338. German. 3.Alstadhaug KB, Kvarenes HW, Prytz J, Vedeler C. A case of relapsing-remitting facial palsy and ipsilateral brachial plexopathy caused by HSV-1. J Clin Virol. 2016 May;78:62-5. Ukrainian. 4.Tang YW, Cleavinger PJ, Li H, et al. Analysis of candidate-host immunogenetic determinants in herpes simplex virus-associated Mollaret's meningitis. Clin Infect Dis. 2000 Jan;30(1):176–8. 5.Maltsev DV. [Mannose binding lectin deficiency]. Ukrainskyi tetapevtychnyi zhurnal. 2015; 1:80-9. Ukrainian. 6.Maltsev DV, Hurzhii OO. Toxoplasma chorioretinitis in primary myeloperoxidase MPO deficiency: A case report. J Ophthalmol. (Ukraine). 2019;4:75-81. 7.Krichevskaia GI, Katargina LA. [Viral and non-viral infections in the etiopathogenesis of uveitis]. Vestn Oftalmol. 2020;136(1):124–9. 8.Heiligenhaus A, Klotsche J, Niewerth M, et al. Similarities in clinical course and outcome between juvenile idiopathic arthritis (JIA)-associated and ANA-positive idiopathic anterior uveitis: data from a population-based nationwide study in Germany. Arthritis Res Ther. 2020 Apr 15;22(1):81. 9.Leite JL, Bufalo NE, Santos RB, et al. Herpesvirus type 7 infection may play an important role in individuals with a genetic profile of susceptibility to Graves' disease. Eur J Endocrinol. 2010 Feb;162(2):315-21. 10.Dyachenko P, Dyachenko A., Smiianova O, et al. Identification of human herpesvirus 7 in the cerebrospinal fluid of adult ukrainian with relapsing-remitting multiple sclerosis. A case study. Wiad Lek. 2018; 71(8):1636–8. 11.Nora-Krukle Z, Chapenko S, Logina I, et al. Human herpesvirus 6 and 7 reactivation and disease activity in multiple sclerosis. Medicina (Kaunas). 2011;47(10):527–31. 12.Venâncio P, Brito MJ, Pereira G, Vieira JP. Anti-N-methyl-D-aspartate receptor encephalitis with positive serum antithyroid antibodies, IgM antibodies against mycoplasma pneumoniae and human herpesvirus 7 PCR in the CSF. Pediatr Infect Dis J. 2014 Aug;33(8):882–3. 13.Foldager L, Köhler O, Steffensen R, et al. Bipolar and panic disorders may be associated with hereditary defects in the innate immune system. J Affect Disord. 2014;164:148–54. 14.Friborg JT, Jarrett RF, Koch A, et al. Mannose-binding lectin genotypes and susceptibility to Epstein-Barr virus infection in infancy. Clin Vaccine Immunol. 2010 Sep;17(9):1484-7. 15.Manuel O, Pascual M, Trendelenburg M, Meylan PR. Association between mannose-binding lectin deficiency and cytomegalovirus infection after kidney transplantation. Transplantation. 2007;83(3):359–62. 16.Maltsev DV. [Mannose binding lectin deficiency]. Ukrainskyi medychnyi chasopys. 2015;2 (106):91-6. Ukrainian. 17.Segat L, Crovella S, Comar M, et al. MBL2 gene polymorphisms are correlated with high-risk human papillomavirus infection but not with human papillomavirus-related cervical cancer. Hum Immunol. 2009 Jun; 70(6):436-9. 18.Maggi F, Pifferi M, Michelucci A, et al. Torque teno virus viremia load size in patients with selected congenital defects of innate immunity. Clin Vaccine Immunol. 2011 Apr;18(4):692-4. 19.Tanha N, Troelsen L, From Hermansen ML, et al. MBL2 gene variants coding for mannose-binding lectin deficiency are associated with increased risk of nephritis in Danish patients with systemic lupus erythematosus. Lupus. 2014 Oct;23(11):1105–11. 20.Saevarsdottir S, Vikingsdottir T, Vikingsson A, et al. Low mannose binding lectin predicts poor prognosis in patients with early rheumatoid arthritis. A prospective study. J Rheumatol. 2001 Apr;28(4):728–34. 21.Mumcu G, Inanc N, Aydin SZ, et al. Association of salivary S. mutans colonisation and mannose-binding lectin deficiency with gender in Behçet's disease. Clin Exp Rheumatol. Mar-Apr 2009;27(2 Suppl 53):S32-6. 22.Aydin SZ, Atagunduz P, Inanc N, et al. Mannose binding lectin levels in spondyloarthropathies. J Rheumatol. 2007 Oct; 34(10):2075-7. 23.Potlukova E, Freiberger T, Limanova Z, et al. Association between low levels of Mannan-binding lectin and markers of autoimmune thyroid disease in pregnancy. PLoS One. 2013;8(12):e81755. 24.Ramasawmy R, Spina GS, Fae KC, et al. Association of Mannose-Binding Lectin Gene Polymorphism but Not of Mannose-Binding Serine Protease 2 With Chronic Severe Aortic Regurgitation of Rheumatic Etiology. Clin Vaccine Immunol. 2008;15(6):932–6. 25.Carter S, Xie K, Knight D, et al. Granulomatous Uveitis and Conjunctivitis Due to Common Variable Immune Deficiency: A Case Report. Ocul Immunol Inflamm. 2019;27(7):1124–6. 26.Pasquet F, Kodjikian L, Mura F, et al. Uveitis and common variable immunodeficiency: data from the DEF-I study and literature review. Ocul Immunol Inflamm. 2012 Jun;20(3):163–70. 27.Farhoudi A., Chavoshzadeh Z., Mir Saeid Ghazi B, et al. Recurrent infections and bilateral uveitis in a patient with CD8 deficiency. Iran J Allergy Asthma Immunol. 2005;4(1):43–5. 28.Angioi K, Terrada C, Locatelli A, et al. Ocular Manifestations of X-linked Chronic Granulomatous Disease: About Two Atypical Case Reports. Ocul Immunol Inflamm. 2015;23(6):458–61. 29.Kubicka-Trzaska A. [Immunologic disturbances in patients with idiopathic posterior uveitis]. Klin Oczna. 2000;102(4):253–8. 30.Spârchez M, Lupan I, Delean D, et al. Primary complement and antibody deficiencies in autoimmune rheumatologic diseases with juvenile onset: a prospective study at two centers. Pediatr Rheumatol Online J. 2015 Nov 21;13:51. 31.Miserocchi E, Modorati G. Rituximab for noninfectious uveitis. Dev Ophthalmol. 2012;51:98-109. 32.Valdimarsson H. Infusion of plasma-derived mannan-binding lectin (MBL) into MBL-deficient humans. Biochem Soc Trans. 2003 Aug;31(Pt 4):768-9. The authors declare no conflict of interest which could influence their opinions on the subject or the materials presented in the manuscript.
|