J.ophthalmol.(Ukraine).2019;4:75-81.
|
http://doi.org/10.31288/oftalmolzh201947581 Toxoplasma chorioretinitis in primary myeloperoxidase MPO deficiency: A case report D.V. Maltsev1, Cand Sc (Med); O.O. Hurzhii2, Ophthalmologist, MD 1 Institute of Experimental and Clinical Medicine at the Bogomolets NMU; Kyiv (Ukraine) 2 Ailas Clinic; Kyiv (Ukraine) E-mail: dmaltsev@ukr.net TO CITE THIS ARTICLE: Maltsev DV, Hurzhii OO. Toxoplasma chorioretinitis in primary myeloperoxidase MPO deficiency: A case report. J.ophthalmol.(Ukraine).2019;4:75-81. http://doi.org/10.31288/oftalmolzh201947581
We present a clinical case of primary partial myeloperoxidase (MPO) deficiency in a young female patient with recurrent episodes of opportunistic infections. Initially, she presented to the clinical immunologist (DVM) seeking help with her severe clinical manifestations of chronic fatigue syndrome. Blood polymerase chain reaction (PCR) was positive for Epstein Barr virus. Brain MRI showed signs of external hydrocephalus and bilateral hippocampal sclerosis. After double antiviral therapy with valacyclovir (3 g daily, orally) and recombinant human alpha-2b-interferon (3 mln units every other day intramuscularly) for 3 months, the blood PCR became negative, and clinical symptoms completely resolved. A year later, the patient presented again to the immunologist, but this time due to abruptly decreased vision in her right eye. She was referred to the ophthalmologist. There were ophthalmoscopic and fluorescein angiography (FA) signs of acute chorioretinitis in the right eye. Serological testing revealed increased IgM titers to Toxoplasma gondii. Blood PCR was positive for Toxoplasma gondii. The patient was diagnosed with acute Toxoplasma chorioretinitis based on the clinical-and-laboratory, ophthalmoscopic and FA findings, and was treated for toxoplasmosis with spiramycin (1.5 mln units diluted in 200 ml of 0.9% normal saline, daily for 14 days, intravenously, and thereafter, 1000 mg three times a day after meals, for 14 days, orally). In addition, she received continuous basic immunomodulating therapy with recombinant human interferon gamma (500,000 units every other day at bedtime for 6 months) for compensation of MPO deficiency, as per the national guidelines. This normalized the percentage content and functional activity of MPO in neutrophils, and enabled prevention of further opportunistic infection episodes. This case report demonstrates that treatment of eyes affected by opportunistic infections in immunocompromised patients requires cooperation between ophthalmologists and clinical immunologists. Keywords: chorioretinitis, toxoplasmosis, myeloperoxidase deficiency
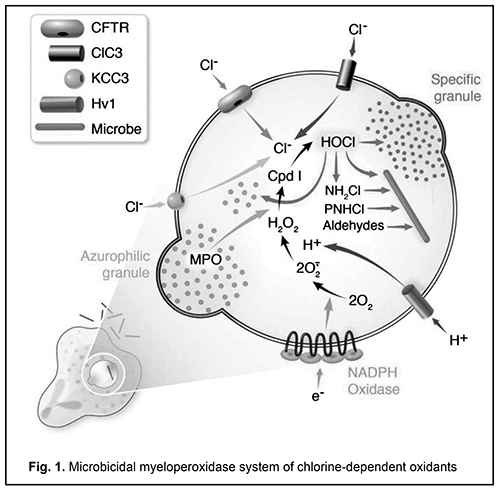
Deficiency of myeloperoxidase (MPO), a disorder of neutrophils, monocytes and macrophages, is a common primary minor immunodeficiency, affecting 1 in 2,000 to 4,000 individuals in the general population in the USA and Europe [1]. MPO deficiency results in a reduced production of hypochlorite anion, a microbicidal agent, with an associated impairment in chlorine-dependent pathogen oxidation in phagolysosomes [2, 3]. Therefore, a reduced microbicidal capacity of phagocytes is characteristic for MPO deficiency, and results in incomplete phagocytosis and immune response evasion by pathogens [4] (Fig. 1).
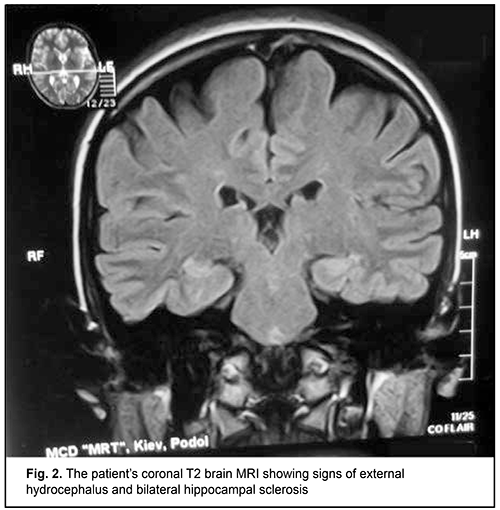
Although this immune deficiency is commonly compensated due to the activity of another microbicidal enzyme, NADPH oxidase, and does not result in severe clinical consequences [4], clinically manifested forms of the disease may occur and require special therapy [5]. Infections caused by opportunistic pathogens [6, 7] and some autoimmune, allergic, immunoinflammatory, and oncologic manifestations [8] have been described in patients with MPO deficiency. Particularly, individuals with such an immunoeficiency are prone to developing frequent attacks of or a severe episode of invasive candidiasis [9, 10]. The capacity of MPO to exert a virucidal effect with cytomegalovirus [11] and human immunodeficiency virus type 1(HIV-1) [12] have been demonstrated, and, therefore, some MPO-deficient patients have reduced resistance to opportunistic viral agents. Such autoimmune syndromes as rheumatic fever [13], multiple sclerosis [4] and rheumatoid arthritis [14] have been reported in MPO deficiency. Vasomotor dysfunction [15], parkinsonism [16], hemostasis hemostasis impairment [17], autism spectrum disorders [18] and impaired biological transformation of medications [19] are considered secondary immune deficiency syndromes, and associated with the pleiotropic nature of the biologic effects of MPO extending well beyond immune system function. A number of diagnostic laboratory assays for verification of MPO deficiency in human phagocytes are available, particularly automated hematography (a screening assay); flow cytofluorometry; biochemical assays including guaiacol peroxidation and alanine decarboxylation assays; spectroscopic analysis; enzyme immunoassay; immunoblotting; cytochemical techniques including benzidine and 4-Chloro-1-naphthol staining; immunocytochemical techniques; luminol-enhanced chemiluminescence of phagocytes; and electron microscopy [2, 20-23]. MPO deficiency should be differentiated from chronic granulomatous disease, neutrophil-specific granule deficiency, type I glycogenosis, hyperimmunoglobulin-E-recurrent infection (Job's) syndrome, Kostman's disease, Ch?diak-Higashi syndrome, adhesion molecule deficiency, and Shwachman syndrome, in which various phases of phagocytosis in neutrophils and monocytes/macrophages are impaired [2]. Primary MPO deficiency shows heterogeneous patterns of the complete deficiency state on the genomic level. The R569W missense mutation is a classic mutation described by Nauseef et al [1]. Nevertheless, Marchetti et al [24] conducted a study to investigate the nature of the mutations underlying the disease of the immune system. The genetic characterization of the subjects showed the presence of 3 already-known mutations (c.752T>C, c.1705C>T and c.1566_1579del14) and 6 novel mutations: four missense mutations (c.995C>T, c.1112A>G, c.1715T>G and c.1927T>C), than a deletion of an adenine within exon 3 (c.325delA) and a mutation within the 3’ splice site of intron 11 (c.2031–2A>C). As per the locally adapted National Evidence-based Guidelines [25] and Unified Clinical Pathway for Providing Primary, Secondary and Tertiary Care [26] in MPO deficiency in phagocytes, antimicrobial medications are administered in addition to basic treatment comprising recombinant human interferon gamma for compensation for immunodeficiency through an increase in the percentage content and functional activity of the affected enzyme in phagocytizing cells [27]. Although toxoplasma is a well-known opportunistic pathogen, until now, there have been no reports on the development of toxoplasmosis in primary MPO deficiency in humans. We present, to our knowledge, the first case of the development of toxoplasmosis in primary MPO deficiency in humans. A 25-year-old woman presented to a clinical immunologist (DVM) with complaints of weakness, fatigue, poor health, joint and muscle pain, and persistent hyperthermia (37.2-37.5 °C). She reported that she had been ill for the past 2 years and that her weakness had been progressive. In addition, the weakness had been severe over the past several months. Her physical examination was remarkable for congested posterior pharyngeal wall; hypertrophy of palatine tonsils; lymphadenopathy of submaxillary, anterior and posterior cervical, and axillary lymphatic nodes; and harsh breathing heard in the inferior portions of the lungs to auscultation. Her complete blood count was remarkable for absolute and relative lymphocytosis, but her erythrocyte sedimentation rate (ESR) was normal. Polymerase chain reaction (PCR) with primers specific for herpes viruses was positive for Epstein Barr virus (100,000 infectious units in a sample). The repeat PCR test demonstrated the same findings. In addition, the patient underwent ultrasonic examination of the thyroid gland, lung X-ray examination, and brain MRI. The results of these examinations, as well as serum pituitary, thyroid and gonad hormone levels were found normal. Conventional brain MRI showed signs of external hydrocephalus (expanded convex spaces and unusually deep sulci and thinned convolutions) and bilateral hippocampal sclerosis, the signs characteristic of infections with lymphotropic herpes viruses (Fig. 2).
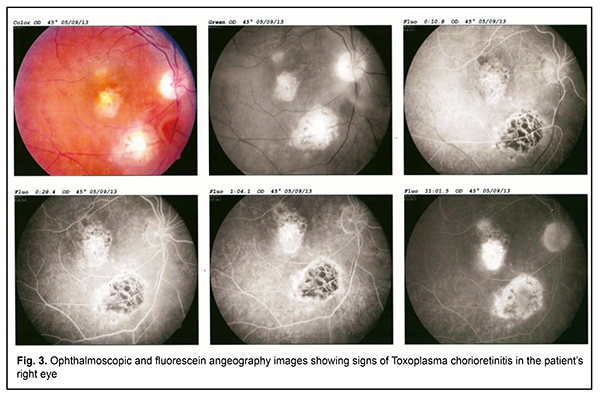
Since no other cause for the clinical symptoms was established, weakness, poor health, and hyperthermia were thought to be associated with reactivated Epstein Barr virus infection. Therefore, the patient was administered double antiviral therapy with an acyclic analogue of guanosine, valacyclovir (3 g daily, orally) and recombinant human alpha-2b-interferon (3 mln units every other day, intramuscularly), in accordance with the findings of a recent controlled clinical study [28, 29]. After two months of this continuous combinatory antiviral therapy, no viral DNA in blood was detected, and, since her condition sharply improved, weakness was almost gone, and body temperature normalized, she was released from the clinic to be supervised by a family doctor. However, a year after her release from the clinic, her condition again worsened. She complained of light flashes in front of her eyes, blinking sensation, floating flies, moving black spots in the field of vision. In addition, decreased visual acuity, mixed conjunctival injection and increased sensitivity to light and tearing were noted. Because of the patient’s complaints, she was referred to the ophthalmologist, where she underwent ophthalmoscopy and fundus fluorescein angiography (FA); there were ophthalmoscopic signs of right eye chorioretinitis (Fig. 3). The right eye ophthalmoscopy found numerous retinal (including macular) foci. Fluorescein angiography showed pathological foci of hyperfluorescence in the macular and inferior temporal areas along vascular arcades in all phases. In addition, we revealed signs of retinal pigment epithelium atrophy. Such an ophthalmoscopic and FA picture of chorioretinitis may accompany an acute infection.
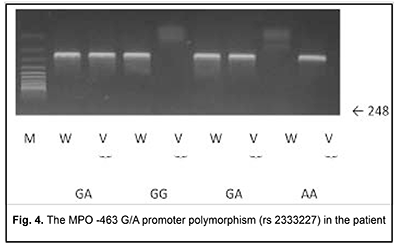
Blood PCR was negative with primers specific for HSV-1, HSV-2, VZV, EBV, CMV, HHV-6, HHV-7, HHV-8; adenovirus; TT virus; enteroviruses; measles; Salmonella typhi; epidemic parotiditis; and Borrelia. However, serological testing revealed increased IgM titers to Toxoplasma gondii. Blood PCR with was positive for Toxoplasma gondii (10,000 infectious units in a sample). The patient was diagnosed with acute Toxoplasma chorioretinitis based on the clinical-and-laboratory, ophthalmoscopic and FA findings. Since there were already two opportunistic infection episodes registered in the patient’s record, she underwent immunological evaluation. Her HIV tests were negative. Immunological evaluation included complete blood count. In addition, lymphocyte subpopulations were studied using laser flow cytometric (Epics XL- Flow Cytometer; Beckman Coulter Inc., Brea, CA) analysis and indirect immunofluorescence with monoclonal antibodies to CD markers (CD3+, CD3+CD4+, CD3+CD8+, CD3–CD19+, CD3–CD16+CD56+, CD3+CD16+CD56+; Beckman Coulter Assays). Phagocytosis was assessed using the latex test; phagocytic number (the number of pathogenic units engulfed by one phagocyte), phagocytic index (percentage of phagocytes involved in phagocytosis), number of active phagocytes and phagocytic capacity of blood (the number of pathogenic units that can be neutralized by the phagocytes contained in 1 liter of blood) were determined. Serum levels of IgM, IgG and IgA were determined by the radial immunodiffusion method of Mancini, whereas those of IgE, IgD, IgG1, IgG2, IgG3 and IgG4, using enzyme immunoassay (Vector Brest, RF). The Nitro Blue Tetrazolium Test was many times conducted at the Zabolotnyi Institute of Microbiology and Virology as well at the Neuroimmunology Laboratory, Institute of Neurosurgery, NAMS of Ukraine, and the results of this test were normal, which is important to differentiate MPO deficiency from a chronic granulomatous disease. MPO deficiency was assessed both quantitatively (with laser flow cytometry;) and qualitatively (with enzyme immunoassay;). These tests demonstrated findings of 44.1% and 11.2 units of 18 units, respectively. The repeat laser flow cytometry and enzyme immunoassay tests demonstrated similar findings (51.6% and 10.5 units, respectively). Therefore, there were quantitative and qualitative deficiencies of MPO in the patient’s phagocytes. In order to differentiate primary MPO deficiency from secondary MPO deficiency, genetic tests were conducted at the Neurobiochemistry Department, Institute of Neurosurgery. The R569W mutation was not detected; however, MPO ?463G/A polymorphism was detected (Fig. 4). This immunodeficiency was believed to be the cause for recurrent episodes of infection by the opportunistic agent in the patient.
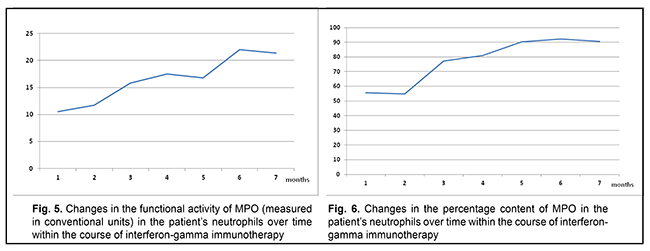
The patient was treated for toxoplasmosis with an antibiotic agent, spiramycin (1.5 mln units diluted in 200 ml of normal saline, daily for 14 days, intravenously, and thereafter, 1000 mg three times a day after meals, for 14 days, orally). Since the patient was found to have a primary MPO deficiency, she received continuous basic immunomodulating therapy with recombinant human interferon gamma (ingaron; 500,000 units every other day at bedtime for 6 months) for compensation for immunodeficiency, as per the national guidelines. Figs. 5 and 6 show changes in the percentage content and functional activity of the affected enzyme in neutrophils over time within the course of immune therapy.
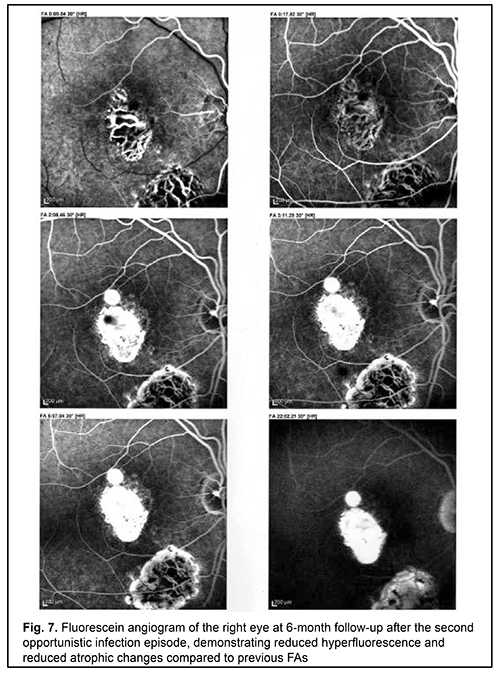
The treatment prevented the progression of toxoplasmosis and contributed to a partial restoration of patient’s vision and prevention of further opportunistic infection episodes. There were no further opportunistic infection episodes over a three-year follow-up. Today the patient is feeling fairly well. Fig. 7 shows the fluorescein angiogram of the right eye at 6-month follow-up after the second opportunistic infection episode, demonstrating reduced hyperfluorescence and reduced atrophic changes compared to previous FAs.
This case report demonstrates that treatment of eyes affected by opportunistic infections requires cooperation between ophthalmologists and clinical immunologists. Such patients should undergo immunological evaluation in order to identify the cause of reactivation of the opportunistic infectious agent. Commonly, this reactivation is a consequence of impaired immune surveillance in the immunocompromised body. Severe opportunistic infections, including those affecting eyes, may develop not only due to secondary immunodeficiency, HIV/AIDS or immunosuppressive medication effect, but also due to primary immunodeficiency. Two general types of immunodeficiency disorders have been reported: (a) classic genetic immunodeficiency disorders (like combined immunodeficiency or Bruton agammaglobulinemia) are uncommon and usually result in severe clinical manifestations developing as early as early childhood, and (b) minor immunodeficiencies are common in population, characterized by a variable course of disease and present with a heterogeneous clinical picture. This case demonstrates that not only classical, but also minor primary immunodeficiencies should be taken into account while searching for the cause of reactivation of the opportunistic infectious agent. Our patient was found to have an MPO deficiency in phagocytes which is attributed to minor immunodeficiencies and common in population. In this regard, the Ministry of Health of Ukraine has adopted the locally adapted guidelines and the unified clinical pathway for providing care to MPO-deficient patients. Identifying the cause of immunodeficiency will enable us not only to explain the development of severe opportunistic infection in the eye care patient, but also to administer specific immunotherapy to compensate for this immunodeficiency, which would be impossible without using immune diagnostics. In conclusion, this case report demonstrates that administering immunotherapy with recombinant human interferon gamma as per the unified clinical pathway enabled (a) to improve the outcome of treatment for Toxoplasma chorioretinitis through restoration of the amount and functional activity of the affected MPO enzyme, and (b) to prevent further opportunistic infection episodes—that is, it showed to be effective in controlling the course of the immune system disease. References 1.Nauseef WM. Diagnostic assays for myeloperoxidase and myeloperoxidase deficiency. Methods Mol Biol. 2014;1124:537-46. 2.Dinauer MC. Disorders of neutrophil function: an overview. Methods Mol Biol. 2014;1124:501-15. 3.Xiao X, Saha P, Yeoh BS, et al. Myeloperoxidase deficiency attenuates systemic and dietary iron-induced adverse effects. J Nutr Biochem. 2018 Dec;62:28-34. 4.Pahwa R, Jialal I. Myeloperoxidase Deficiency. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019-2018 Oct 27. 5.Kazmirchuk VE, Tsaryk VV, Maltsev DV. Myeloperoxidase deficiency – congenital and acquired disorders of neutrophil function. Fundamental and applied sciences today. 2014;3:32–6. 6.Domingues-Ferreira M, Levy A, Barros NC, et al. Case report of myeloperoxidase deficiency associated with disseminated paracoccidioidomycosis and peritoneal tuberculosis. Rev Soc Bras Med Trop. 2017 Jul-Aug;50(4):568-570. 7.Grossl NA, Candel AG, Shrit A, Schumacher HR. Myeloperoxidase deficiency and severe sepsis. South Med J. 1993 Jul;86(7):832-6. 8.Ohno H. Association of primary myeloperoxidase deficiency and myeloproliferative neoplasm. Intern Med. 2010;49(22):2527-8. 9.Kalinski T, Jentsch-Ullrich K, Fill S, et al. Lethal candida sepsis associated with myeloperoxidase deficiency and pre-eclampsia. APMIS. 2007 Jul;115(7):875-80. 10.Milligan KL, Mann D, Rump A, et al. Complete Myeloperoxidase Deficiency: Beware the "False-Positive" Dihydrorhodamine Oxidation. J Pediatr. 2016 Sep;176:204-6. 11.El Messaoudi K, Verheyden AM, Thiry L, et al. Human recombinant myeloperoxidase antiviral activity on cytomegalovirus. J Med Virol. 2002 Feb;66(2):218-23. 12.Chochola J, Yamaguchi Y, Moguilevsky N, et al. Virucidal effect of myeloperoxidase on human immunodeficiency virus type 1-infected T cells. Antimicrob Agents Chemother. 1994 May;38(5):969-72. 13.Pat?ro?lu T, G?ng?r HE, Belohradsky JS, et al. Myeloperoxidase deficiency: the secret under the flag of unstained cell. Turk J Haematol. 2013 Jun; 30(2): 232–3. 14.Bell AL, Markey GM, Alexander HD, et al. Myeloperoxidase deficiency in a patient with rheumatoid arthritis: oxygenation and radical activity by phagocytic cells. Br J Rheumatol. Br J Rheumatol. 1993 Feb;32(2):162-5. 15.Rudolph TK, Wipper S, Reiter B. et al. Myeloperoxidase deficiency preserves vasomotor function in humans. Eur Heart J. 2012 Jul;33(13):1625-34. 16.Kazimirchuk VE, Maltsev DV, Slobodin TN, Golovchenko IuI. [Parkinson's syndrome in young women with myeloperoxidase deficiency of phagocytes]. Mezhdunarodnyi Nevrologicheskii Zhurnal. 2011; 1(39):15-24. Russian. 17.d'Onofrio G, Mancini R, Vallone R, et al. Acquired neutrophil myeloperoxidase deficiency: an indicator of subclinical activation of blood coagulation? Blood Cells. 1983;9(3):455-66. 18.Russo AJ, Krigsman A, Jepson B, Wakefield A. Low serum myeloperoxidase in autistic children with gastrointestinal disease. Clin Exp Gastroenterol. 2009;2:85-94. 19.Kettle AJ, Winterbourn CC. Superoxide-dependent hydroxylation by myeloperoxidase. J Biol Chem. 1994 Jun 24;269(25):17146-51. 20.Albrett AM, Ashby LV, Dickerhof N, et al. Heterogeneity of hypochlorous acid production in individual neutrophil phagosomes revealed by a rhodamine-based probe. J Biol Chem. 2018 Oct 5;293(40):15715-15724. 21.Azarova LA, Vasilenko LP, Oblamskaia GV. [Hereditary myeloperoxidase deficiency: its diagnosis on the Technicon H.1 hematological analyzer]. Gematol. Transfuziol.1993 Feb;38(2):41-2. Russian. 22.Becker R, Pfl?ger KH. Myeloperoxidase deficiency: an epidemiological study and flow-cytometric detection of other granular enzymes in myeloperoxidase-deficient subjects. Ann Hematol. 1994 Oct;69(4):199-203. 23.Ro?niak-B?k K, Jele? A, B?k M. Analysis of Separation of White Blood Cells in Peripheral Blood as an Indicator of MPO Deficiency. Ann Clin Lab Sci. 2017 Aug;47(4):422-431. 24.Marchetti C, Patriarca P, Solero GP, et al. Genetic studies on myeloperoxidase deficiency in Italy. Jpn J Infect Dis. 2004 Oct;57(5):S10-2. 25.[Locally Adapted National Evidence-based Guidelines for Managing Myeloperoxidase Deficiency. State Expert Center of the Ministry of Health of Ukraine and Ukrainian Society of Specialists for Immunology, Allergology and Immunorehabilitation]. Ukrainian. Available at http://mtd.dec.gov.ua/images/dodatki/2016_609_DefMiel_Fago/2016_609_AKN_... 26.[Unified Clinical Pathway for Providing Care to Patients with Myeloperoxidase Deficiency]. 2016. Ukrainian. Available at http://mtd.dec.gov.ua/images/dodatki/2016_609_DefMiel_Fago/2016_609_YKPM... 27.Maltsev DV. [Efficacy of long-term continuous immunomodulating therapy with recombinant human interferon gamma for patients with clinically manifested forms of myeloperoxidase deficiency]. Immunologiia I allergologiia. 2015; 1:44-53. Russian. 28.Maltsev DV. 2015. The effectiveness of combined antiviral therapy in chronic mononucleosis caused by Epstein-Barr virus. European EBV meeting. Karolinska Institutet. Stockholm. p. 33. 29.Ramsaransing G, Teelken A, Prokopenko VM. Low leucocyte myeloperoxidase activity in patients with multiple sclerosis. J Neurol. Neurosurg Psychiatry. 2003;74(7):953-5. The authors certify that they have no conflicts of interest in the subject matter or materials discussed in this manuscript.
|