J.ophthalmol.(Ukraine).2022;2:65-67.
|
http://doi.org/10.31288/oftalmolzh202226567 Received: 13 January 2021; Published on-line: 30 April 2022 Primary mesodermal dysgenesis of the cornea (Peter´s anomaly) Leopoldo Garduño Vieyra, Dr.; Dr. Raúl Rúa Martínez, Dr.; Francisco Javier Rodríguez Hernández, Dr.; Isabel De la Fuente Batta, Dr. Clínica Oftalmología Garduño; Irapuato, Guanajuato (México) E-mail: ruamartinez@yahoo.es TO CITE THIS ARTICLE: Leopoldo Garduño Vieyra, Raúl Rúa Martínez, Francisco Javier Rodríguez Hernández, Isabel De la Fuente Batta. Primary mesodermal dysgenesis of the cornea (Peter´s anomaly) . J.ophthalmol.(Ukraine).2022;2:65-7. http://doi.org/10.31288/oftalmolzh202226567
Primary mesodermal dysgenesis of the cornea, also known as Peter´s anomaly (PA) or keratolenticular dysgenesis, is a rare congenital eye condition caused by an abnormal development of the anterior segment. PA is characterized by unilateral or bilateral corneal opacity (leucoma), that appears since the early neonatal period. The incidence of PA in the United States of America is approximately 1.5 per 100,000 live births. PA is known as Peters-plus syndrome when it presents with systemic malformations. In this article we describe the clinical presentation of an 18-year-old patient with PA that arrived to medical consultation due to bilateral corneal opacities since birth. The patient´s parents refer that he has hypoacusis and deny other systemic pathologies. Clinical exploration reveals a visual acuity of 20/30 of the right eye and 20/100 of the left eye. According to the clinical findings and the absence of systemic anomalies, the patient was diagnosed with PA type II. Key words: сongenital, Peter´s аnomaly, leucoma, cataract
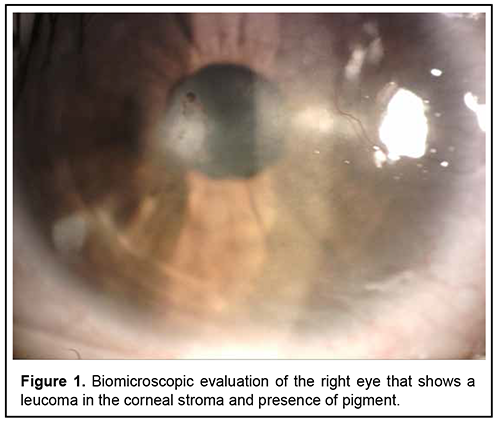
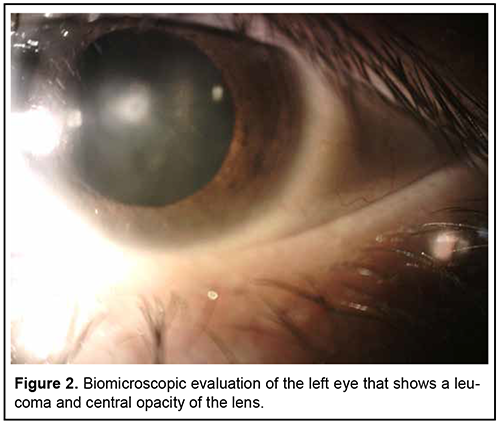
Introduction Primary mesodermal dysgenesis of the cornea, also known as Peter´s anomaly (PA) or keratolenticular dysgenesis, is a rare congenital eye condition caused by an abnormal development of the anterior segment. This anomaly was first described by Albert Peters, a German ophthalmologist. PA is characterized by unilateral or bilateral corneal opacity (leucoma), that appears since the early neonatal period [1, 4]. This anomaly can be associated with other ocular anomalies, such as: glaucoma, cataract, microphthalmia, optic nerve hypoplasia, aphakia, aniridia, and retinal anomalies [1, 8]. Histologically, Iridocorneal adhesions appear at the edge of the opacity, and Descemet membrane and endothelium are absent in the area of the opacity [5]. PA can be divided into two subtypes: type I (central corneal stromal opacity associated with iridocorneal adhesions, with no lens involvement) and type II (presence of keratolenticular adhesions and/or cataract) [3]. PA is known as Peters-plus syndrome when it presents with systemic malformations, like mental retardation, cleft lip and palate, shorter limbs, short stature, abnormal ears, and developmental delay. This syndrome is associated with homozygous mutations in the B3GLCT gene [2, 4, 7]. Brain imaging findings associated with PA are mainly corpus callosum abnormalities and malformations of cortical development. Other less common reported abnormalities are hypoplasia of the cerebellar vermis, absent septum pellucidum, hippocampus abnormalities and cerebral calcification [8]. The incidence of PA varies around the world. In North China, congenital corneal opacities (CCO) represent 13% of the pediatric keratoplasties. In India, 12% of corneal transplantations in children were for CCO. In Spain, the incidence of CCO was reported as 3.11 out of 100,000 births. In the United States of America, PA represents de 65% of corneal transplants in infants and its incidence is approximately 1.5 per 100,000 live births [5, 7]. Most cases are sporadic, but autosomal dominant and recessive inheritance have also been reported. Several gene mutations have been reported in PA, some of the implicated genes are FOXC1, FOXE3, PAX6, PITX2/RIEG1, and CYP1B1 [4, 7]. Faber et al recently described that a mutation in the COL4A1 gene, which encodes for collagen Ivα1, may be associated with PA [4]. These genes are involved in the differentiation and migration of neural crest migration to the posterior cornea. The central corneal opacity is formed by the incomplete separation of the corneal epithelium and the lens vesicle. The failure in the migration of neural crest cells into the anterior segment is also involved [10]. Treatment of PA depends on the size, localization, laterality, and associated anomalies. Treatment options are observation, penetrating keratoplasty, controlling glaucoma and cataract surgery [1, 7]. This study aimed to describe the clinical presentation, ophthalmologic evaluation findings, additional studies and treatment of an 18-year-old patient with PA. Case report An 18-year-old patient arrives for ophthalmologic consultation due to bilateral congenital corneal opacities without diagnosis. The patient reported that he had begun to have blurred vision for 5 years. That´s why he received medical attention at other health centers. In these centers, he was treated with unspecified topical medication without improvement. A history of ocular inflammation or other symptoms could not be specified. The patient´s parents refer that he has hypoacusis and deny other systemic pathologies. Clinical exploration reveals a visual acuity of 20/30 of the right eye and 20/100 of the left eye. Biomicroscopic evaluation shows right eye with a leucoma in the corneal stroma that extended to Descemet's membrane and endothelium, presence of pigment (figure 1), deep anterior chamber angle; the rest of the anterior segment within normal limits. Left eye had a leucoma that compromises Descemet's membrane and endothelium, deep anterior chamber angle and opacity of the lens (figure 2). Fundus examination was normal in both eyes. No signs of keratolenticular adhesion were found.
According to the clinical findings and the absence of systemic anomalies, PA type II was suspected and proper genetical studies were solicitated. The clinical diagnosis was confirmed by the genetical studies that revealed a FOXC1 gene mutation. Observation was decided as treatment due to the actual visual acuity. Periodic follow up was established to evaluate complications such as glaucoma and to program a cataract surgery when required. Discussion In the case reported in this article, the patient presents with bilateral congenital corneal opacities localized in the stroma. He also presented a central opacity of the lens in the left eye. These findings are consistent with PA type II. Peter´s plus was discarded because the patient didn´t present systemic anomalies, and he had a normal growth and development. PA must be differentiated from other CCO. Also, other ocular and systemic anomalies must be inquired. CCO is a term that comprise a group of diseases of the eye that are present at birth and cause loss of transparency of the cornea. Incidence of CCO is 2.2 to 3.11 per 100,000 births. It is more frequent that the alteration is bilateral. These alterations occur between de 6th and 16th weeks of life during the differentiation of the anterior segment. Several factors, alone or in combination, may disrupt this process. Some of them are infectious, genetic, metabolic, developmental, traumatic, toxic, and idiopathic. An early diagnosis is important, because proper treatment must be established to reduce the risk of amblyopia and lifelong visual impairment. The most frequent etiology of CCO is PA [5]. The differential diagnosis of PA includes iridocorneal endothelial syndrome, posterior polymorphous dystrophy, Axenfeld-Rieger anomaly and syndrome, congenital hereditary endothelial dystrophy, and congenital hereditary stromal dystrophy. Iridocorneal endothelial syndrome involves three clinical entities: progressive essential iris atrophy, Cogan-Reese syndrome, and Chandler syndrome. This syndrome presents with abnormalities of the corneal endothelium, progressive obliteration of the corneal angle, and iris anomalies (atrophy and polycoria). These changes lead to corneal decompensation and secondary glaucoma. Posterior polymorphous dystrophy presents with iridocorneal adhesions, membranes, and ectropion uveae. This condition, differing from iridocorneal endothelial syndrome, has a genetic component and is rarely progressive [9]. Other differential diagnosis is Axenfeld–Rieger syndrome, characterized by Rieger Anomaly (iris hypoplasia resulting in pseudopolycoria and corectopia) and Axenfeld Anomaly (anteriorization of Schwalbe’s line of the cornea with iris adhesions) [10]. Conclusions CCO are a group of diseases which diagnosis and treatment must be made early, because they have a high risk of developing amblyopia and lifelong visual impairment. PA is the most common cause of CCO. The origin of this anomaly is an abnormal differentiation and migration of the neural crest cells into the anterior segment of the eye. PA manifests in the neonatal period with unilateral or bilateral corneal opacity. Glaucoma, cataract, aniridia and other ocular anomalies may accompany the leucoma. The definite treatment is a penetrating keratoplasty, but complications such as glaucoma and cataract may also receive prompt care.
Acknowledgments: To all the staff of our clinic Disclaimers: The authors remove any responsibility from the journal in relation to the content of this article. Source(s) of support: The authors didn´t received extra monetary support for the realization of this article. Disclosure of relationships and activities: The authors declare no conflict of interests. References 1.Brobova N, Romanova T, Tronina SA. Successful Two-Stage Rehabilitation of The Ectodermal form of Peters Anomaly in Frakcaro-Schmid Syndrome. Ukr J Ophthalmol. 2011;6:49–52. 2.Demir GÜ, Lafcı NG, Doğan ÖA, Kiper PÖŞ, Utine GE. Peters plus syndrome: A recognizable clinical entity. Turk J Pediatr. 2020;62(1):136–40. 3.Elbaz U, Strungaru H, Mireskandari K, Stephens D, Ali A. Long-Term Visual Outcomes and Clinical Course of Patients With Peters Anomaly. Cornea. 2020;00(00):1–9. 4.Faber H, Puk O, Holz A, Biskup S, Voykov B. Identification of a New Genetic Mutation Associated With Peters Anomaly. Cornea. 2020;00(00):1–4. 5.Karadag R, Rapuano CJ, Hammersmith KM, Nagra PK. Causes of congenital corneal opacities and their management in a tertiary care center. Arq Bras Oftalmol. 2020;83(2):98–102. 6.Kurilec JM, Zaidman GW. Incidence of peters anomaly and congenital corneal opacities interfering with vision in the united states. Cornea. 2014;33(8):848–50. 7.Li Y, Zhang J, Dai Y, Fan Y, Xu J. Novel Mutations in COL6A3 That Associated With Peters’ Anomaly Caused Abnormal Intracellular Protein Retention and Decreased Cellular Resistance to Oxidative Stress. Front Cell Dev Biol. 2020;8(November):1–13. 8.Samara A, Eldaya RW. Ocular and brain imaging findings in Peters’ anomaly: A case report and literature review. Radiol Case Reports. 2020;15(7):863–6. 9.Walkden A, Au L. Iridocorneal endothelial syndrome: Clinical perspectives. Clin Ophthalmol. 2018;12:657–64. 10.Weigele J, Bohnsack BL. Genetics underlying the interactions between neural crest cells and eye development. J Dev Biol. 2020;8(4):1–24.
Authors approve the manuscript and agree with what is expressed in it and do not have conflict of interest in this article.
Received 19.01.2021
|