J.ophthalmol.(Ukraine).2021;5:71-76.
|
http://doi.org/10.31288/oftalmolzh202157176 Received: 12 April 2021; Published on-line: 23 October 2021 Bilateral morning glory syndrome associated with persistent fetal vasculature syndrome and corpus callosum agenesis: a case report N. F. Bobrova, T. V. Romanova, A. V. Shilik SI "The Filatov Institute of Eye Diseases and Tissue Therapy of the NAMS of Ukraine"; Odesa (Ukraine) E-mail: filatov.detskoe7@gmail.com TO CITE THIS ARTICLE: Bobrova NF, Romanova TV, Shilik AV. Bilateral morning glory syndrome associated with persistent fetal vasculature syndrome and corpus callosum agenesis: a case report. J.ophthalmol.(Ukraine).2021;5:71-6. http://doi.org/10.31288/oftalmolzh202157176
Background: The morning glory (MG) syndrome is a rare, sporadic and commonly unilateral anomaly. The MG and persistent fetal vasculature (PVF) syndromes are generally considered as isolated ocular manifestations of failures in various stages of embryogenesis of the eye. Purpose: To describe the clinical manifestations of a bilateral combined ocular pathology, the MG syndrome and PVF syndrome, associated with congenital central nervous system (CNS) and bone anomalies. Material and Methods: We described a rare variant of binocular manifestation of the MG syndrome associated both with the PVF syndrome and CNS involvement in a 7-month infant. Results: In the pediatric case reported here, the bilateral combination of MG syndrome and PVF syndrome is accompanied by congenital CNS anomalies, corpus callosum agenesis and vicarious ventriculomegaly. To the best of our knowledge, this congenital association has been not reported previously, which made us to report this case. Conclusion: Morning glory syndrome is a rare congenital disorder whose pathogenesis is still not fully understood. The rare infant case reported here demonstrates an association of the MG syndrome with another congenital anomaly of the eye (the persistent fetal vasculature syndrome) and a major congenital anomaly of the CNS, corpus callosum agenesis, which requires not only detailed and comprehensive medical evaluation, but also longitudinal patient surveillance in cooperation with allied specialties like neuropathology, pediatrics, etc. Keywords: morning glory syndrome, persistent fetal vasculature syndrome, congenital CNS and bone anomalies
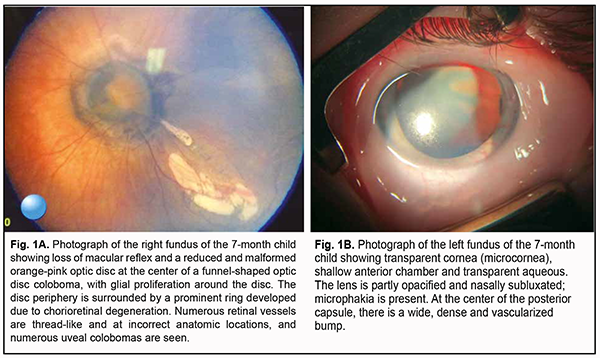
Introduction The morning glory (MG) syndrome is a rare, sporadic and commonly unilateral anomaly [1, 2]. This disorder is twice as common in women as is in men [3] and can be associated with various central nervous system (CNS) anomalies [4, 5]. The persistent fetal vasculature (PVF) syndrome results from failure of the vascular lens capsule, primary vitreous and hyaloid vasculature to regress [6-8]. The MG and PVF syndromes are generally considered as isolated ocular manifestations of failures in various stages of embryogenesis of the eye. Here we report a rare variant of binocular manifestation of the MG syndrome associated both with the PVF syndrome and central nervous system (CNS) involvement. Our aim was to describe the clinical manifestations of a bilateral combined ocular pathology, the MG syndrome and PVF syndrome, associated with congenital CNS and bone anomalies. Results The parents presented their 7-month boy to the Pediatric Ophthalmology Department of the Filatov institute with the complaint of vision loss more in the left eye than in the right eye and bilateral nystagmus on February 9, 2021. The infant had been the product of his mother’s fifth pregnancy, which was complicated by acute respiratory viral infection during the 11th week of gestation, and born in a spontaneous vaginal delivery at the 40th week, with a birth weight of 4,000 g. He had no significant family history. Immediately after birth, microphthalmia of the left eye was noted. A comprehensive eye examination was performed under general anesthesia. The infant was examined by the geneticist, and ophthalmologist. TORCH screen was positive for rubella IgG (11.21 IU/ml against a norm of 0-10 IU/ml). Biomicroscopy and ophthalmoscopy data Right eye: The eye was quiescent and showed nystagmus. An 11.5 mm-diameter cornea was transparent. The anterior chamber was uniformly deep, and the aqueous was transparent. A round pupil was central and dilated uniformly; strands of the persistent pupillary membrane (PPM) were seen in the inferior pupil. The lens and vitreous were transparent. Dilated fundus examination found no macular reflex and a reduced and malformed orange-pink optic disc at the center of a funnel-shaped optic disc coloboma, with glial proliferation around the disc. The disc was surrounded by a prominent ring developed due to chorioretinal degeneration. Numerous retinal vessels were thread-like and at the incorrect anatomic locations, and numerous uveal colobomas were seen (Fig. 1A).
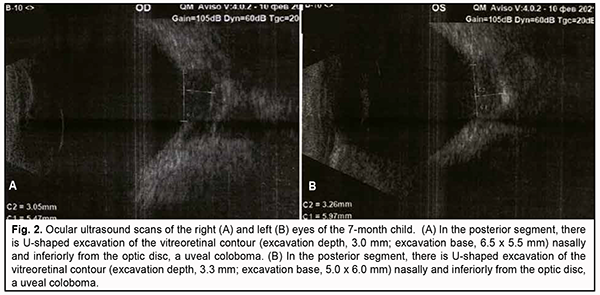
Left eye: The eye was quiescent, reduced in size, and showed nystagmus. There was a transparent cornea (microcornea) with a diameter of 10.0 mm. The anterior chamber was uniformly deep, and the aqueous was transparent. A round pupil was central and dilated uniformly. There lens had an irregularly hazy appearance and was nasally subluxated; microphakia was present. At the center of the posterior capsule, there was a wide, dense and vascularized bump (Fig. 1B) with the hyaloid artery extending from it. On ophthalmoscopy, the fundus was cloudy, and no macular reflex was seen. Similarly to the right eye, there was a funnel-shaped optic disc coloboma, and the disc was surrounded by a prominent ring developed due to chorioretinal degeneration. Retinal vessels were thread-like and at the incorrect anatomic locations. Visual acuity measured by Teller acuity cards was 0.1 OD and 0.01 OS. Intraocular pressure (IOP) was 20.0 mmHg OD and 14.0 mmHg OS. A-scan ultrasound biometry showed an axial length of 20.23 mm OD and 18.29 mm OS. Ultrasound biomicroscopy (UBM, AVISO V:4.0.2, Quantel Medical, France) data Right eye: The anterior segment was sonographically unremarkable. There was U-shaped excavation of the vitreoretinal contour (excavation depth, 3.0 mm; dimensions, 6.5 x 5.5 mm) at the optic disc in the posterior eye, a uveal coloboma (Fig. 2A).
Left eye: Ultrasound biomicroscopy of the left eye demonstrated a sonographically small globe, a shallow anterior chamber, and a non-homogeneous and nasally displaced lens of 5.4-mm thickness and 6.6-mm diameter. The ciliary body appeared attached. The vitreous exhibited individual punctal-and-fibrous structures of low echogenicity. The retina appeared attached. There was excavation of the vitreoretinal contour (excavation depth, 3.3 mm; dimensions, 5.0 x 6.0 mm) at the optic disc in the posterior eye, a uveal coloboma (Fig. 2B). Brain and orbital magnetic resonance imaging (MRI) The corpus callosum was not seen, and the lateral ventriculi were located wide apart from each other. Myelination of the white matter was not yet complete. No focal pathological changes were observed in the intraorbital structures. The left lens appeared medially displaced and malformed. The optic nerves were symmetric; the optic nerve width was 0.42 cm OD and 0.33 cm OS (the perineurium of the optic nerve OS was not clearly visualized). The left half of the chiasm was thinned. Brain midline structures were not displaced. The width of the lateral ventricles was as wide as 0.4 cm, and of the third ventricle, 0.7 cm. No pathological changes were seen in the craneovertebral junction. Cerebellar tonsils were normally located. Pneumatization of the paranasal sinuses was not complete and the degree of paranasal sinus pneumatization corresponded to the child’s age. It was concluded that there was MRI evidence of congenital malformation (corpus callosum agenesis), left optic nerve atrophy, and the left lens was medially displaced and malformed. The patients was diagnosed with a “bilateral congenital ocular malformation of the eye, the morning glory syndrome combined with the persistent fetal vasculature syndrome; persistent pupillary membrane in the right eye, and microphthalmia and microcornea complicated by subluxated partial cataract and remnant hyaloid artery in the left eye” based upon a comprehensive diagnostic examination. The secondary diagnosis was congenital malformations of the CNS (corpus callosum agenesis and mild vicarious ventriculomegaly) and bones (“gothic” palate and dental anomaly) and nasolabial fistula. Discussion Reis [9] was the first to describe the uncommon optic disc anomaly now known as morning glory syndrome in 1908, and Handmann [10] reported on 6 patients with the anomaly in 1929. In 1970, Kindler [11] first coined the term morning glory syndrome because the ophthalmoscopic appearance of this anomaly closely resembles the appearance of the South American Ipomoea species commonly named morning glory flower. Most cases of morning glory syndrome are unilateral [1, 12]. The right eye is more frequently involved in unilateral cases (60%). A family history of morning glory syndrome is uncommon. A case of trisomy 4q with a unilateral morning glory disc anomaly has been reported [12]. There have been isolated reports of bilateral morning glory anomaly, the authors of which supposed that the anomaly is of a hereditary origin. Others have reported that the morning glory syndrome was associated with changes in the eye, such as pupillary defect, ciliary cyst, congenital cataract, posterior lenticonus, aniridia, epiretinal fibrosis, strabismus, and lid hemangioma. Findings of visual function studies may include visual field defects like enlarged blind spot and/or central scotoma [13-15]. The embryogenesis of the MG syndrome is not certain. Some researchers believe that the syndrome is a phenotypic manifestation of colobomatous defects (i.e., those related to the embryonic fissure), while some others believe a primary mesodermal defect underpins the development of the MG syndrome. Demp¬ster and colleagues [16] attempted to reconcile these two views by proposing that the basic defect is mesodermal but that some clinical features of the defect may result from a dynamic disturbance between the relative growth of mesoderm and ectoderm. Pollock [17] has proposed that the initial embryologic defect leading to the development of the morning glory disc anomaly is an abnormal funnel-shaped enlargement of the distal optic stalk at its junction with the primitive optic vesicle. When this occurs, invagination of the optic vesicle leads to formation of the embryonic fissure that extends from the newly formed optic cup into the expanded distal optic stalk. Closure of the embryonic fissure occurs in weeks 5 to 6 of the embryonic development. Closure of the embryonic fissure occurs normally, but because of the increased dimensions of the distal optic stalk, this process of closure fails to obliterate the space within the distal stalk, resulting in a persistent excavated defect at the site of entry of the optic nerve into the eye. According to this hypothesis, glial and vascular abnormalities develop later in embryogenesis and reflect abnormal development of mesodermal elements in a setting of primary neuroectodermal dysgenesis. The persistent fetal vasculature syndrome is a congenital anomaly of the eye that was first described by Collins in 1908 [18]. A delay in the regression of the hyaloid artery and embryonic vascular lens capsule underpins the development of the syndrome. Goldberg [8] proposed to use a unified term, the persistent fetal vasculature syndrome (PVFS) rather than previously used denominations of the syndrome (retrolental fibroplasia, persistent hyperplastic lens tissue, persistent vascular lens capsule, persistent hyaloid artery, etc.) in 1997. PFVS has been regarded as primarily unilateral in approximately 90% of patients and affects men and women at equal rates. It usually occurs as a nonheritable, unilateral eye disorder. Regression of the hyaloid artery begins in the fourth month of embryonic development, and is completed by the time of birth, leaving Cloquet’s canal that runs through the vitreous body from the optic disc to the posterior surface of the lens. The clinical manifestations of the syndrome vary and depend on whether a particular portion of the primary vitreous preserved or not. E.g., in the case reported here, an iris anomaly in the form of persistent pupillary membrane was found in the better eye. Less common are large arterial vessels extending from the optic disc anteriorly to the posterior lens capsule (and sometimes anastomosing with the iris vasculature), a so called remnant hyaloid artery. The classical signs of a marked PFVS are microphthalmia, shallow anterior chamber, persistent pupillary membrane, cataract and vascularized retrolental membrane, the latter causing traction of the ciliary processes that extend centrally and become visible once the pupil is dilated [19-21]. Cataract is an early complication of PFVS and is detected practically in all children with the syndrome, with predominance (86.9%) of complicated cataract with posterior synechiae, heterogeneously dense anterior capsule and hazy posterior capsule [1, 6, 19, 22]. It has been reported that MS syndrome can be associated with PFVS and Peters syndrome. Azuma and colleagues [23] supposed that these three syndromes can be inherited. The PAX6 gene is involved in ocular morphogenesis and is expressed in the developing central nervous system and numerous ocular tissues during development [23, 24]. PAX6 mutations have been detected by Azuma and colleagues [6] in various ocular anomalies, including Peters’ anomaly, congenital cataracts, MG syndrome and PFVS [23]. A histopathological study reported on the combination of MG syndrome, PFVS, Peters’ anomaly, congenital cataract, total retinal detachment, and crescent-shaped retinal fold and supposed that these associations may be accidental or a manifestation of neural crest-shaped mesoectodermal anomaly [25]. Cennamo and colleagues [26] reported on a case of MS syndrome associated with PFVS and lens colobomas, and supported the unitary hypothesis that MS syndrome, optic pit, and optic nerve colobomas are expressions of a single pathogenic process. Fei and colleagues [27] revealed the coexistence of PFVS in a significant percentage of patients with MS syndrome, suggesting a potential common genetic link. Matsubara and colleagues [25] reported on two cases of anterior staphyloma associated with Peters’ anomaly and PFVS. In the pediatric case reported here, MG syndrome manifested practically similarly in the fundus of both eyes. There was, however, MRI evidence of changes in the orbital portion of the left optic nerve with the development of optic atrophy. In addition, PFVS manifested by mild PPM in the better-seeing right eye and by microphthalmia and microcornea complicated by subluxated partial cataract and remnant hyaloid artery in the worse-seeing left eye. In MG syndrome, systemic congenital disorders are mostly related to brain development anomalies [28], and can be found in 45% of cases. The MG anomaly is closely associated with basal encephalocele [4]. Transsphenoidal encephalocele is a congenital malformation in which a meningeal pouch, often containing the chiasm and adjacent hypothalamus, protrudes inferiorly through a defect in the sphenoid bone. Sometimes children with this basal meningocele have a midline cleft in the soft palate, and oral extension of meningocele can be visualized. Commonly they have a wide head, a flat nose, and a midline notch or cleft in the upper lip. In the presence of meningocele protruding to the nasopharynx, there may be difficulty with nasal respiration due to nasal blockage, leading to mouth breathing and snoring. Because the hypothalamus can be contained within the encephalocele, hypopituitarism can occur, affecting growth hormone and antidiuretic hormone production [29]. Other cerebrovascular anomalies have also been frequently demonstrated in the MG syndrome, most notably ipsilateral intracranial vascular dysgenesis. If both intracranial carotid arteries are involved, then hypertrophy of collateral vessels at the base of the brain can occur, resembling a “puff of smoke” on arteriogram [30]. This descriptively is called moyamoya syndrome [31, 32]. Some patients with morning glory disc anomaly, infantile hemangiomas, and dysplasia of the carotid vasculature have PHACE syndrome [25], which includes posterior fossa malformations, facial hemangiomas, arterial anomalies, cardiac anomalies and aortic coarctation. Cases of MG syndrome associated with congenital kidney anomalies and CHARGE syndrome have been reported. The association of MG syndrome with hypophyseal nanism has been rarely reported. Hypophyseal nanism develops in patients with MG syndrome secondary to compression of the pituitary gland by a basal encephalocele, leading to secondary pituitary hypertrophy. Other midline intracranial anomalies have been reported, including agenesis of the corpus callosum, and absent optic chiasm, as well as an association with renal agenesis. The corpus callosum is a thick band of nerve fibers that divides the cerebrum into left and tight hemispheres (Fig. 3). It connects the left and right sides of the brain, allowing for communication between both cerebral hemispheres. The corpus callosum transfers motor, sensory, and cognitive information between the brain hemispheres and also involved in several functions of the body including communication between brain hemispheres, eye movement, maintaining the balance of arousal and attention, and tactile localization. The corpus callosum plays an important role in vision by combining the separate halves of our visual field, which process images separately in each hemisphere. It also allows us to identify the objects we see by connecting the visual cortex with the language centers of the brain. In addition, the corpus callosum transfers tactile information (processed in the parietal lobes) between the brain hemispheres.
Corpus callosum agenesis (CCA) is a birth defect in which the corpus callosum is partially or completely absent. The corpus callosum normally develops between the 12th and 20th weeks of embryonic development and continues to undergo changes in its structure through childhood, adolescence and even adulthood. CCA can be caused by several factors, including chromosomal mutations, intrauterine infections, drug and toxic effects on the fetus, and cyst-induced abnormal development of the brain. Individuals with CCA may exhibit delayed cognitive development. Other potential problems include hearing deficits, distorted head or facial features, spasms, and seizures [2]. Researchers have discovered that the resting-state brain activity in both those with healthy brains and those with CCA look essentially the same. This indicates that the brain compensates for the missing corpus callosum by rewiring itself and establishing new nerve connections between the brain hemispheres. The actual process involved in establishing this communication is still unknown. In the pediatric case reported here, the bilateral combination of MG syndrome and PFVS is accompanied by congenital CNS anomalies, corpus callosum agenesis and vicarious ventriculomegaly. To the best of our knowledge, this congenital association has been not reported previously, which made us report this case. There is no universally agreed standard approach to treating combined MG syndrome, PFVS and congenital CNS anomalies. Lee and Traboulsi [33] believe that it is essential to stabilize visual acuity in order to prevent the development of amblyopia. Detailed fundus evaluation should be performed longitudinally to detect serious sequelae of MS syndrome, such as serous retinal detachment that may originate in the peripapillary area and extend through the posterior pole [34]. Surgery for transsphenoidal encephalocele may be too difficult to perform and is considered by many authorities to be contraindicated, because herniated brain tissue may include vital structures, such as the hypothalamic–pituitary system, optic nerves and chiasm, and anterior cerebral arteries [34]. We believe that surgical repair of PFVS through the removal of subluxated cataract in the case reported here is contraindicated now because of poor visual prognosis, given MRI evidence of left optic nerve atrophy). In addition, microphthalmia and microphakia make IOL implantation impossible, whereas the surgery itself may affect the further growth of the eye. Of note is also that prolonged general anesthesia is a concern in apparent CNS pathology. Longitudinal patient surveillance in collaboration with other specialties (including neuropathology, pediatrics, rehabilitology, etc.) is advisable. Conclusion Morning glory syndrome is a rare congenital disorder whose pathogenesis is still not fully understood. The rare infant case reported here demonstrates an association of the MG syndrome with another congenital anomaly of the eye (the persistent fetal vasculature syndrome) and a major congenital anomaly of the CNS, corpus callosum agenesis, which requires not only detailed and comprehensive medical evaluation, but also longitudinal patient surveillance in cooperation with allied specialities (neuropathology, pediatrics, etc.). Patients with the MG syndrome require MRI of the brain and orbits to determine if any congenital CNS anomaly is present. In addition, if possible, MRI angiography should be performed to exclude any associated congenital vascular anomaly (moyamoya syndrome, PHACE, etc) that would expose the patient to the risk of cerebrovascular ischemia. A pediatric ophthalmologist should be alert when performing the diagnostic workup of congenital anomalies of the eye. Referring such children to the Pediatric Ophthalmology Department of the Filatov institute for verifying the diagnosis and determining the further treatment strategy is believed to be reasonable.
References 1. Bobrova NF, Vit VV. [Atlas of congenital and hereditary disease of the eye]. Odessa: Palmira; 2006. p.68-9. Russian. 2. Tyszka JM, Kennedy DP, Adolphs E, Paul LK. Intact Bilateral Resting-State Networks in the Absence of the Corpus Callosum. J Neurosci. 2011 Oct 19; 31(42): 15154–162. 3. Brown GC, Tasmand WS. Congenital Anomalies of the Optic Disc. New York, NY: Grune & Stratton;1983. p. 123-70. 4. Ellika S, Robson CD, Heidary G, Paldino MG. Morning glory disc anomaly: characteristic MR imaging findings. Am J Neuroradiol. 2013 Oct;34(10):2010-4. 5. Strominger MB. Morning Glory Syndrome. Neuro-Ophthalmology. 2015 Oct 14. 6. Bobrova NF, Skripnichenko ZM. [Toxic, congenital, and secondary cataracts]. Odessa: Feniks; 2017. Russian. 7. Birnholz JC, Farrell EE. Fetal hyaloid artery: timing of regression with US. Radiology. 1988 Mar;166(3):781-3. 8. Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV). Am J Ophthalmol. 1997 Nov;124(5):587-626. 9. Reis W. Eine wenig bekannte typische Missbildung am Sehenrveheintritt: Unschriebene Grubenbildung auf der Papilla n. optici. Z Augenheilkd. 1908;19:505–28. 10. Handmann M. Erbliche vermutlich angeborene zentrale gliose Entartung des Sehnerven mit besonderer Beteilingung der Zentralgefaesse. Klin Monatsbl Augenheilkd. 1929;83:145-52. 11. Kindler P. Morning glory syndrome: unusual congenital optic disk anomaly. Am J Ophthalmol. 1970 Mar;69(3):376-84. 12. Miller NR, Newman NJ, eds. Anomalies of the optic disc. In: The essentials: Walsh & Hoyt's Clinical Neuroophthalmology. 5th ed. Baltimore: Williams & Wilkins. p.117-23. 13. Safari A, Jafari E, Borhani-Haghighi A. Morning glory syndrome associated with multiple sclerosis. Iran J Neurol. 2014 Jul 4;13(3):177-80. 14. Steinkuller PG. The morning glory disk anomaly: Case report and literature review. J Pediatr Ophthalmol Strabismus. Mar-Apr 1980;17(2):81-7. 15. Traboulsi EL, Jurdi-Nuwayhid F, Torbey NS, et al. Aniridia, atypical iris defects, optic pit and the morning glory disc anomaly in a family. Ophthalmic Paediatr Genet. 1986 Aug;7(2):131-5. 16. Dempster AG, Lee WR, Forrester JV, McCreath GT. The 'morning glory syndrome' - a mesodermal defect? Ophthalmologica. 1983;187:222-30. 17. Pollock S. The morning glory disc anomaly: contractile movement, classification, and embryogenesis. Doc Ophthalmol. 1987 Apr;65(4):439-60. 18. Collins ET. Developmental deformities of the crystalline lens. Ophthalmoscope. 1908; 6:577–83, 663. 19. Khvatova AV, Sudovskaia TV. [Clinical features, diagnostic and treatment options for unilateral congenital cataracts associated with persistent primary vitreous syndrome: a textbook for physicians]. Moscow; 2002. Russian. 20. Antely I, Cohen E, Karshai E, BenEzra D. Unilateral persistent hyperplastic primary vitreous: course and outcome. J AAPOS. 2002 Apr;6(2):92-9. Crossref PubMed 21. Kanigowska K, Grałek M, Chipczyńiska B, Hautz W. [Problems in surgical management of persistent hyperplastic primary vitreous in children]. Klin Oczna. 2006;108(1-3):51-4. Polish. 22. Roche O, Sylla FK, Beby F, et al. [Persistence and hyperplasia of primary vitreous]. J Fr Ophtalmol. 2007 Jun;30(6):647-57. 23. Azuma N, Yamaguchi Y et.al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003 Jun;72(6):1565–70. 24. Krasnovid TA, Grubnik NP. [Morning glory syndrome associated with persistent hyaloid artery]. Oftalmol Zh. 2011;6:53-5. Russian. 25. Matsubara A, Ozeki H, Matsunaga N, et al. Histopathological examination of two cases of anterior staphyloma associated with Peters’ anomaly and persistent hyperplastic primary vitreous. Br J Ophthalmol. 2001 Dec;85(12):1421–5. 26. Cenamo G, Liguori G, Pezone A, Laccarino G. Morning glory syndrome associated with marked persistent hyperplastic primary vitreous and lens colobomas. Br J Ophthalmol. 1989 Aug;73(8):684–6. 27. Fei P, Zhang Q, Li J, Zhao P. Clinical characteristics and treatment of 22 eyes of morning glory syndrome associated with persistent hyperplastic primary vitreous. Br J Ophthalmol. 2013 Oct;97(10):1262–7. 28. Xu LT, Grigorian AP. Morning Glory anomaly. Neuro-Ophthalmology. 2020;5. 29. Loddenkemper T, Friedman NR, Ruggieri PM, et al. Pituitary stalk duplication in association with moya moya disease and bilateral morning glory disc anomaly. J Neurol. 2008 Jun;255(6):885-90. 30. Quah BL, Hamilton J, Blaser S, et al. Morning glory disc anomaly, midline cranial defects and abnormal carotid circulation: an association worth looking for. Pediatr Radiol. 2005 May; 35(5):525-8. 31. Ponnatapura J. Morning glory syndrome with Moyamoya disease: A rare association with role of imaging. Indian J Radiol Imaging. Apr-Jun 2018;28(2):165-8. 32. Komiyama M, Yasui T, Sakamoto H, et al. Basal meningoencephalocele, anomaly of optic disc and panhypopituitarism in association with moyamoya disease. Pediatr Neurosurg. 2000 Aug;33(2):100-4. 33. Lee BJ, Traboulsi EI. Update on the morning glory disc anomaly. Ophthalmic Genet. 2008 Jun;29(2):47-52. 34. Brodsky MC. Pediatric Neuro-Ophthalmology. 2nd ed. New York: Springer; 2010.
|