J.ophthalmol.(Ukraine).2018;4:32-38.
|
https://doi.org/10.31288/oftalmolzh201843236 Received: 25 May 2018; Published on-line: 31 August 2018 Clinical efficacy of treatment for subclinical-stage axial diabetic optic neuropathy M.A. Karliychuk,1 Cand Sc (Med), P.A. Bezditko,2 Dr Sc (Med), Prof. Received: 25 May 2018 Published: 31 August 2018 1 Higher State Educational Establishment of Ukraine «Bukovinian State Medical University»; Chernivtsi (Ukraine) 2 Kharkiv National Medical University; Kharkiv (Ukraine) E-mail: mari13karli@gmail.com TO CITE THIS ARTICLE: Karliychuk MA, Bezditko PA. Clinical efficacy of treatment for subclinical-stage axial diabetic optic neuropathy. J.ophthalmol.(Ukraine).2018;4:32-38. https://doi.org/10.31288/oftalmolzh201843236
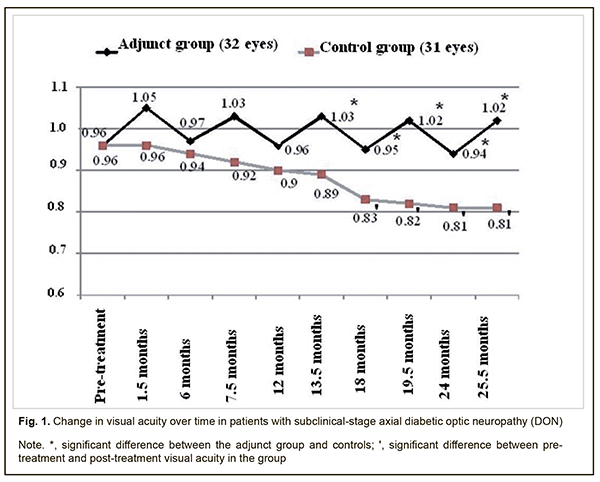
Background: There is no standard of therapy for the treatment of diabetic optic neuropathy (DON). Purpose: To assess the clinical efficacy of thioctic acid, topical brimonidine tartrate and a combination of vitamins В1, В6, and В12 in the management of subclinical-stage axial DON. Materials and Methods: Forty patients (63 eyes) were followed up after being diagnosed with subclinical-stage axial DON. The adjunct group was composed of 20 patients (32 eyes) who were administered (1) thioctic acid (Berlithion) at a dose of one 300 mg tablet a day for 42 days, (2) a 2 ml intramuscular injection of Milgamma, once per 3 days for 21 days, followed by switching to oral regimen, 1 tablet per 3 days for 21 days in two courses during a year, and (3) topical brimonidine tartrate 0.2%, 1-2 drops twice a day on a constant basis, as adjunctive to hypoglycemic therapy. The control group (20 patients; 31 eyes) received hypoglycemic therapy only. In addition to routine eye examination, retinal and optic nerve optical coherence tomography and electrophysiology were performed. Patients underwent an examination at baseline, 1.5 months, 6 months, 7.5 months, 13.5 months, 24 months and 25.5 months after treatment. Results: No progression of optic nerve damage was found in all 32 affected eyes of the adjunct group versus no progression, progression to mild-stage axial DON, and progression to advanced-stage axial DON in 64.5% (20 eyes), 35.5% (11 eyes) and 6.5% (2 eyes), respectively, of the controls at the 25.5-month time point. We found that our treatment attenuated the progression of optic nerve damage in subclinical-stage axial DON, with 25.9% better visual acuity, 29.5 % lower electrically evoked phosphene thresholds, 69.3% less ganglion cell complex (GCC) focal loss volume (FLV), and 29.8% less thickness of the cribriform plate compared to controls at 25.5 months. Conclusion: Our treatment was found to be clinically efficacious in attenuating the progression of optic nerve damage in subclinical-stage axial DON. Keywords: axial diabetic optic neuropathy, subclinical stage, treatment, efficacy Introduction There is no standard of therapy for the treatment of diabetic optic neuropathy (DON). Because DON is a manifestation of diabetic polyneuropathy (DPN), it is reasonable to correct the abnormality with the agents targeted at the pathogenetic components of DPN. Standard therapy for DPN includes measures aimed at achieving and maintaining stable compensation of diabetes mellitus (DM), B group vitamins, and alpha-lipoic acid (ALA, also called thioctic acid) [1-5]. B group vitamins exert metabolic effect on transport in peripheral nerve fibers and their myelination. Neurotropic vitamins serve as coenzymes in various biochemical reactions, improve nerve cell energy, facilitate optimal glucose uptake, enhance neurotrophic protection and regenerative capacity of the peripheral nerves, are synergistic with ALA, and provide support for the involvement of the latter in neuronal metabolism [6-8]. Benfotiamine and pyridoxine, the two substances of Milgamma (Woerwag Pharma, Böblingen, Germany), have the ability to inhibit the formation of advanced glicated end products (AGEs) that can cause cell dysfunction, inflammatory processes and vascular wall disorders. In addition, some authors [9] believe these substances to be neurotrophic inhibitors of AGEs. It has been proved that inhibiting AGEs formation slows the progression of retinal damage in DM [9]. The efficacy of Milgamma in the management of DPN has been proved in a number of randomized placebo-controlled studies [6]. Studies on the mechanisms underlying the effect of thioctic acid on patients with DPN found that the acid contributes to normalization of axonal transport and endoneurial circulation, reduction of oxidative stress, and improvement of vascular dysfunction [2, 4, 5, 10]. As retinal ganglion cell (RGC) apoptosis underlies DON, it deems reasonable to utilize neuroprotectors in the management of the disorder. According to the main conclusions of the preliminary statistical analyses of the EUROCONDOR clinical trial, topical treatment with brimonidine tartrate 0.2% is effective in arresting the progression of neurodegeneration in diabetic patients in whom some degree of neurodegeneration is already present [11]. These data suggest that topical brimonidine tartrate 0.2% is a reasonable option for use in DON. The efficacy of differential treatment for DON in patients with different types and stages of the disease is still to be determined. The purpose of the study was to assess the clinical efficacy of thioctic acid, topical brimonidine tartrate 0.2% and a combination of vitamins В1, В6, and В12 in the management of subclinical-stage axial DON. Materials and Methods Forty patients (63 eyes) were followed up after being diagnosed with subclinical-stage axial DON based on the findings of retinal and optic nerve OCT in accordance with the clinical and diagnostic criteria [12] and classification of DON [13] that we have previously developed. Binocular optic neuropathy was found in 23 persons, and 17 of these were found to have mild-stage axial DON in the fellow eye. The adjunct group was composed of 20 patients (32 eyes) who were administered (1) thioctic acid (Berlithion, Berlin-Chemie, Germany) at a dose of one 300 mg tablet a day for 42 days, (2) a 2 ml intramuscular injection of Milgamma, once per 3 days for 21 days, followed by switching to oral regimen, 1 tablet per 3 days for 21 days in two courses during a year, and (3) topical brimonidine tartrate 0.2%, 1-2 drops twice a day on a constant basis, as adjunctive to hypoglycemic therapy. The control group (20 patients; 31 eyes) received hypoglycemic therapy only. Patients underwent an examination at baseline, 1.5 months, 6 months, 7.5 months, 13.5 months, 24 months and 25.5 months after treatment. In addition to routine eye examination (including visual acuity, tonometry, and dilated ophthalmoscopy), retinal and optic nerve OCT and electrophysiology were performed. Focal loss volume (FLV), a ganglion cell complex (GCC) parameter, was assessed as the integral of deviation in areas of significant focal GCC loss based on GCC measurements with a RTVue-100 OCT system (Optovue, Inc., Fremont, CA). Software programs LC_Thickness_programm.m and main_low_noise_filters_programm.m were used to assess cribriform plate thickness based on OCT measurements [14]. Electrically evoked phosphene thresholds (EPT) were measured to evaluate retinal sensitivity, and critical flicker fusion thresholds (CFFT) were assessed using electric phosphene stimulation with a KNSO-2 apparatus (FOSFEN, Odesa, Ukraine). Means (M), standard deviations (σ), standard errors of the mean (SEM) were calculated using Microsoft Excell 2000. In addition, coefficients of variation (Сν), significance (p), 95% confidence intervals (CI) were calculated. The level of significance p ≤ 0.05 was assumed. Results and Discussion No progression of optic nerve damage was found in all 32 affected eyes of the adjunct group versus no progression, progression to mild-stage axial DON, and progression to advanced-stage axial DON in 64.5% (20 eyes), 35.5% (11 eyes) and 6.5% (2 eyes), respectively, of the controls at the 25.5-month time point. In the adjunct group, the mean visual acuities at 1.5 months, 6 months, 7.5 months, 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 0.97±0.11, 1.03±0.11, 0.96±011, 1.03±0.12, 0.95±0.12, 1.02±0.11, 0.94±0.11 and 1.02±0.11, respectively, and were not significantly different from baseline (0.96±0.12; р>0.05 Fig. 1). In the control group, the mean visual acuities at 1.5 months, 6 months, 7.5 months, 12 months, and 13.5 months, were 0.96±0.11, 0.94±0.10, 0.92±0.09, 0.90±0.09, and 0.89±0.09, respectively, and were not significantly different from baseline (0.81±0.07; р>0.05), whereas at 18 months, 19.5 months, 24 months, and 25.5 months were 0.83±0.08, 0.82±0.07, 0.81±0.07, and 0.81±0.07, and were significantly different from baseline (р<0.05).
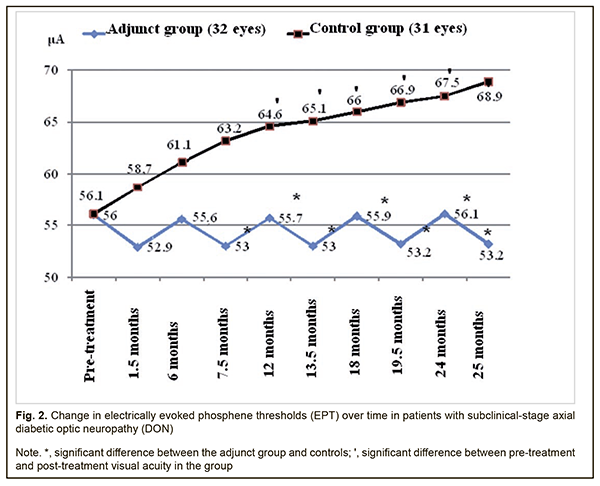
Therefore, compared to the controls, mean visual acuities in the adjunct group at 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 15.7%, 14.5%, 24.4%, 16.0%, and 25.9% higher (p < 0.05). However, there were no significant differences in visual acuity between groups at 1.5 months, 6 months, 7.5 months and 12 months (p > 0.05). In the adjunct group, the mean EPT values at 1.5 months, 6 months, 7.5 months, 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 52.9±4.9 μA, 55.6±6.0 μA, 53.0±5.1 μA, 55.7±6.1 μA, 53.0±5.3 μA, 55.9±6.0 μA, 53.2±5.2 μA, 56.1±5.7 μA, and 53.2±5.1 μA, respectively, and were not significantly different from baseline (56.0±6.1 μA, р > 0.05; Fig. 2). In the control group, the mean EPT values at 1.5 months, 6 months, and 7.5 months were 58.7±6.0 μA, 61.1±6.6 μA, and 63.2±6.5 μA, respectively, and were not significantly different from baseline (56.1±6.2 μA; р>0.05), whereas at 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 64.6±6.0 μA, 65.1±6.0 μA, 66.0±6.1 μA, 66.9±6.2 μA, 67.5±6.4 μA and 68.9±6.6 μA, and were 15.2%, 16.0%, 17.6%, 19.3%, 20.3%, and 22.8%, respectively, higher compared to baseline (р < 0.05).
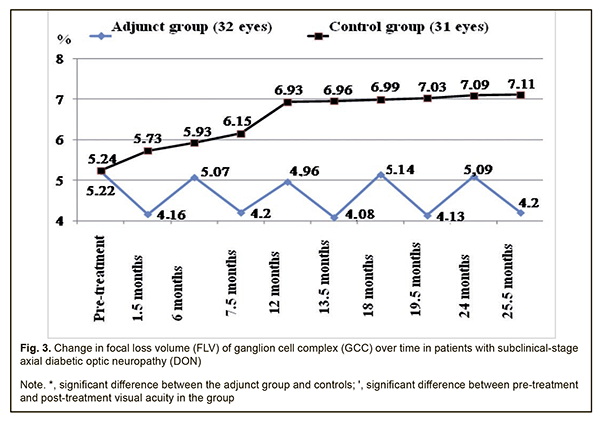
Therefore, compared to the controls, the mean EPT values in the adjunct group at 7.5 months, 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 19.2%, 16%, 22.8%, 18.1%, 25.8%, 20.3% and 25.9%, respectively, lower (p < 0.05). There were, however, no significant differences in EPT between groups at 1.5 months and 6 months (p > 0.05). The mean GCC FLV values in the adjunct group at 1.5 months, 6 months, 7.5 months, 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 4.16±1.43%, 5.07±1.87%, 4.20±1.47%, 4.96±1.93%, 4.08±1.56%, 5.14±1.87%, 4.13±1.63%, 5.09±1.78%, and 4.20±1.38%, respectively, and were not significantly different from baseline (5.22±2.10%, р > 0.05; Fig. 3). In the control group, the mean GCC FLV values at 1.5 months, 6 months, and 7.5 months were 5.73±2.17%, 5.93±2.12%, and 6.15±2.03%, respectively, and were not significantly different from baseline (5.24±2.11%; р>0.05), whereas at 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 6.93±2.12%, 6.96±2.13%, 6.99±2.13%, 7.03±2.16%, 7.09±2.17% and 7.11±2.19%, respectively, and were 32.3%, 32.8%, 33.4%, 34.2%, 35.3%, and 35.7%, respectively, higher compared to baseline (р < 0.05).
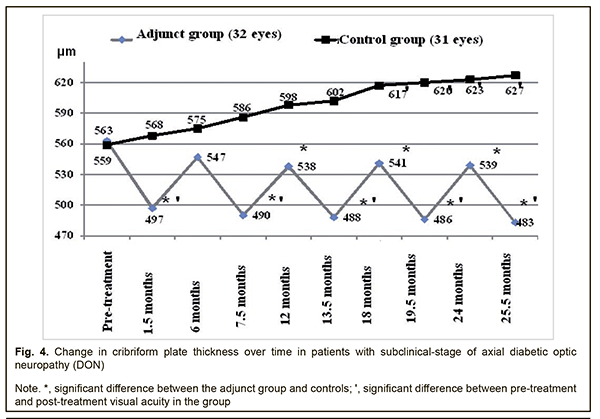
Therefore, compared to the controls, the mean GCC FLV values in the adjunct group at 7.5 months, 12 months, 13.5 months, 18 months, 19.5 months, 24 months, and 25.5 months were 46.4%, 39.7%, 70.6%, 36.0%, 70.2%, 39.3% and 69.3%, respectively, lower (p < 0.05). There were, however, no significant differences in GCC FLV between groups at 1.5 months and 6 months (p > 0.05). The mean thickness of the cribriform plate in the adjunct group at 1.5 months, 7.5 months, 13.5 months, 19.5 months, and 25.5 months was 497±59 μm, 490±62 μm, 488±60 μm, 486±61 μm and 483±60 μm, respectively, which was 13.3%, 14.9%, 15.4%, 15.8%, and 16.6%, lower, respectively, compared to baseline (563±63 μm), and the differences were significant (р<0.05). However, at 6 months, 12 months, 18 months, and 24 months, the mean thickness of the cribriform plate in the adjunct group was 547±61 μm, 538±59 μm, 541±62 μm, and 539±59 μm, respectively, and the differences were not significant compared to baseline (Fig. 4). In the control group, the mean thickness of the cribriform plate at 1.5 months, 6 months, 7.5 months, 12 months, and 13.5 months, was 568±60 μm, 575±61 μm, 586±59 μm, 598±61 μm, 602±58 μm, respectively, and were not significantly different from baseline (559±60 μm; р>0.05), whereas at 18 months, 19.5 months, 24 months, and 25.5 months was 617±55 μm, 620±54 μm, 623±53 μm and 627±56 μm, respectively, which was 10.4%, 10.9%, 11.4%, and 12.2%, respectively, higher compared to baseline (р < 0.05).
Therefore, compared to the controls, the mean thickness of the cribriform plate in the adjunct group at 1.5 months, 7.5 months, 12 months, 13.5 months, 18 months, 19.5 months, 24 months and 25.5 months was 14.3%, 19.6%, 11.2%, 23.4%, 14.0%, 27.6%, 15.6% and 29.8%, respectively, lower (p < 0.05). There was, however, no significant difference in mean thickness of the cribriform plate between groups at 6 months (p > 0.05). In the current study we found that our treatment attenuated the progression of optic nerve damage in subclinical-stage axial DON, with 25.9% better visual acuity, 29.5 % lower electrically evoked phosphene thresholds, 69.3% less GCC FLV, and 29.8% less thickness of the cribriform plate compared to controls at 25.5 months. The results of our treatment with Berlithion for subclinical-stage axial DON are in agreement with the findings of Zhaboiedov et al [15], who developed a “Method of treatment for diabetic neuropathy” involving intravenous infusions supplying intravenous infusions supplying a daily dose of 12 ml (300 mg) of Berlithion in two treatment courses lasting 3-4 days each, followed by switching to oral regimen, 300 mg daily of Berlithion for 2 months. We believe that the absence of a post-treatment increase in the thickness of the cribriform plate in our adjunct group, unlike in controls may be explained by the following: benfotiamine and pyridoxine, the two substances of Milgamma, inhibit the formation of AGEs in collagen, while the accumulation of AGEs in the cribriform plate in DM results in collagen cross-linking and changes in the biomechanical characteristics of the plate, with an increase in its stiffness and reduction in its elasticity [6, 16-18]. Given the intimate contact between the cribriform plate and GCC axons, ultrastructural changes in the plate will result in primary damage to the optic nerve and in secondary damage due to alterations in blood supply and axoplasmic blood flow [16, 19]. Brimonidine tartrate is a highly selective α2-adrenergic agonist that was introduced in 1996 for glaucoma treatment. Moreover, experimental evidence has demonstrated that brimonidine is a potential optic nerve and retinal neuroprotective agent [20], exerts a multifactorial neuroprotective agent, and improves retinal ganglion cell survival in the presence of glutamate, oxidative stress and hypoxia [20]. The agent, however, has been found not effective in preventing neurodegeneration in diabetic patients in the absence of pre-treatment neurodegenerative changes in the GCC [11], which confirms the appropriateness of using it to treat patients diagnosed with DON. The findings of the current study give evidence that the detection of DON at the subclinical stage followed by timely administration of adequate therapy attenuates the progression of disease, and may provide the basis for sight saving in a considerable body of patients with the disease. Conclusion
Administering (1) thioctic acid (Berlithion) at a dose of one 300 mg tablet a day for 42 days, (2) a 2 ml intramuscular injection of Milgamma, once per 3 days for 21 days, followed by switching to oral regimen, 1 tablet per 3 days for 21 days in two courses during a year, and (3) topical brimonidine tartrate 0.2%, 1-2 drops twice a day on a constant basis, as adjunctive to hypoglycemic therapy in patients with sibclinical-stage axial DON, was found to attenuate the progression of optic nerve damage, with 25.9% better visual acuity, 29.5 % lower electrically evoked phosphene thresholds, 69.3% less GCC FLV, and 29.8% less thickness of the cribriform plate compared to controls at 25.5 months. References
|